

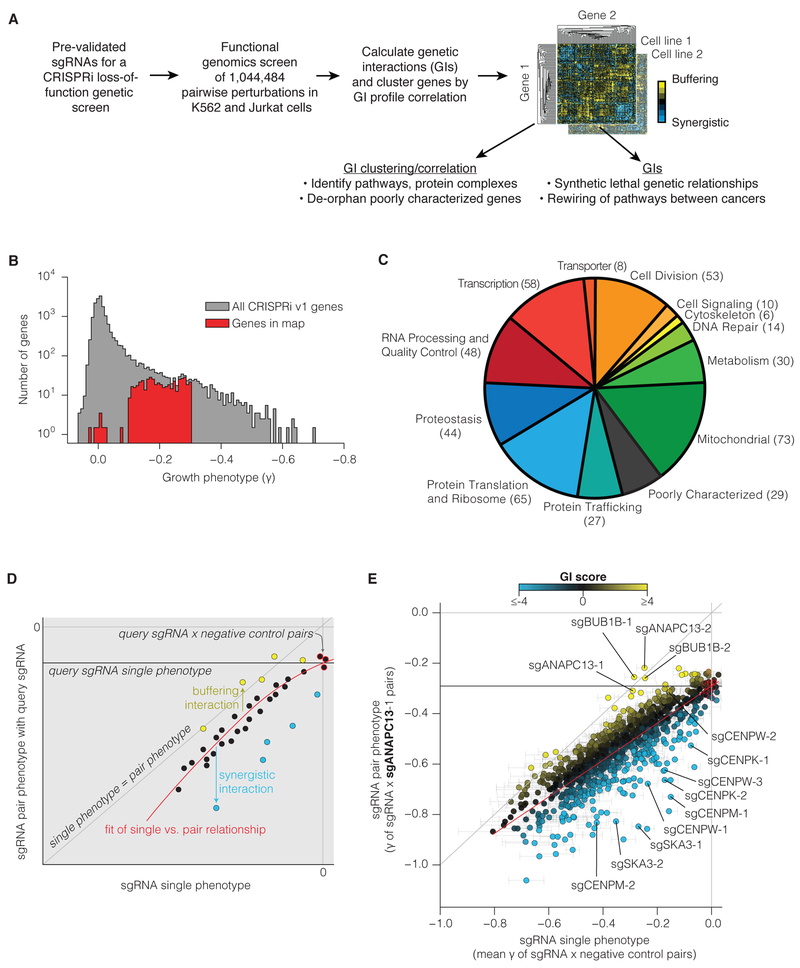
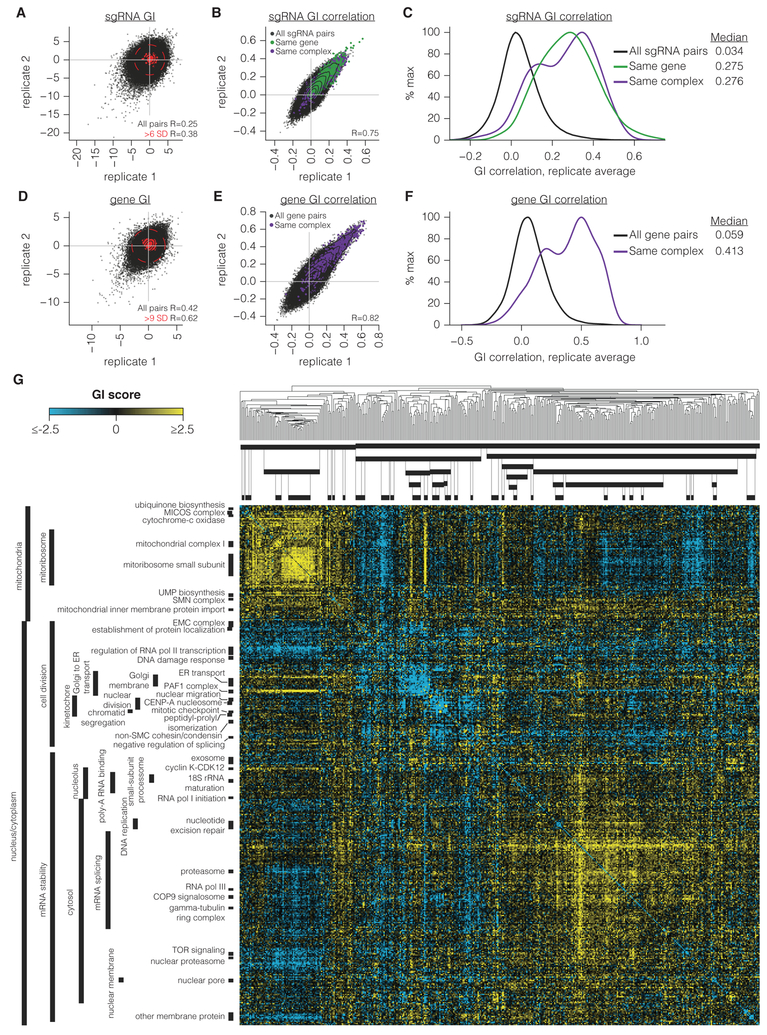
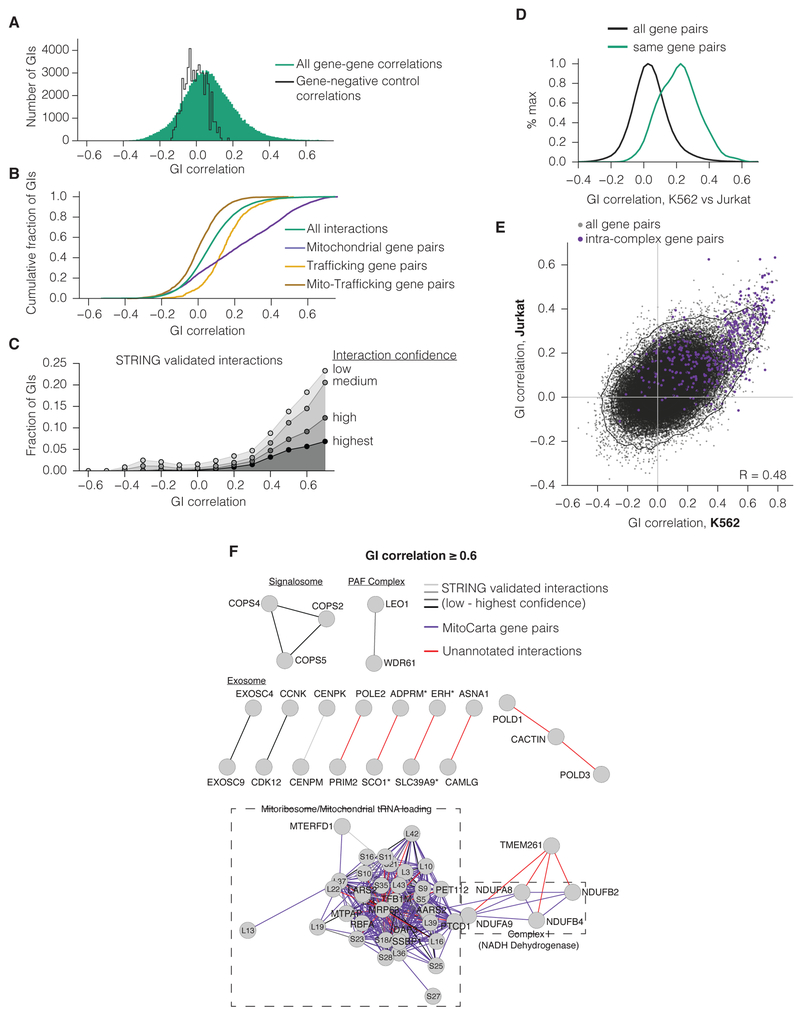
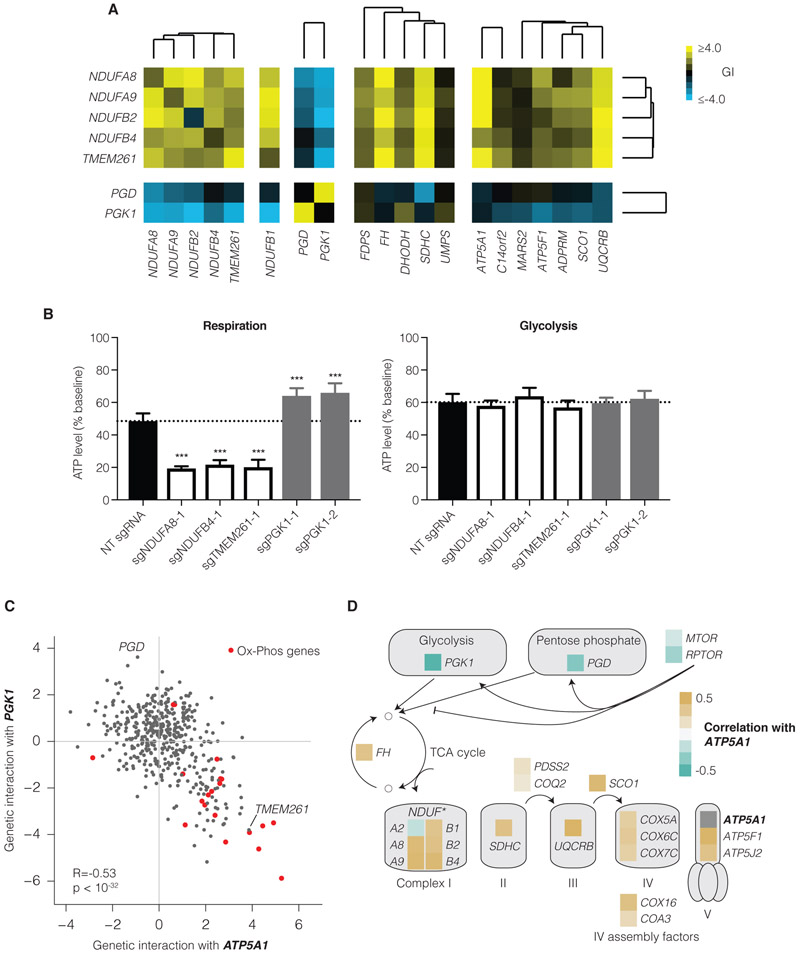
Mapping the Genetic Landscape of Human Cells
- PMID: 30033366
- PMCID: PMC6426455
- DOI: 10.1016/j.cell.2018.06.010
Mapping the Genetic Landscape of Human Cells
Abstract
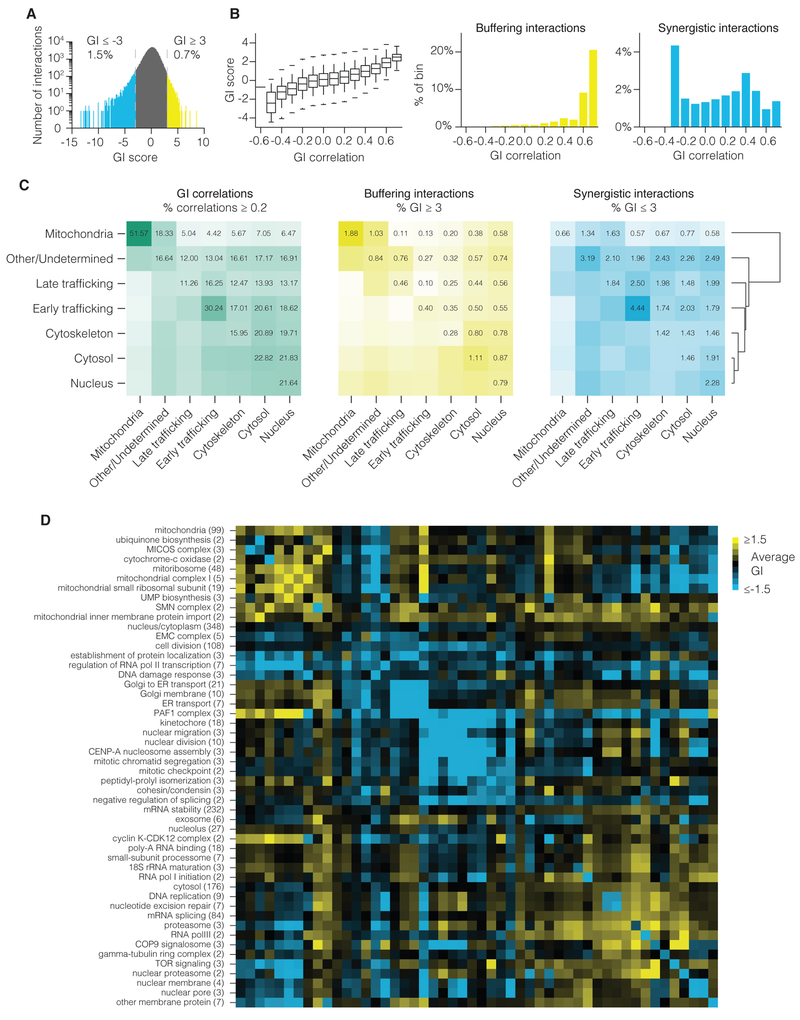
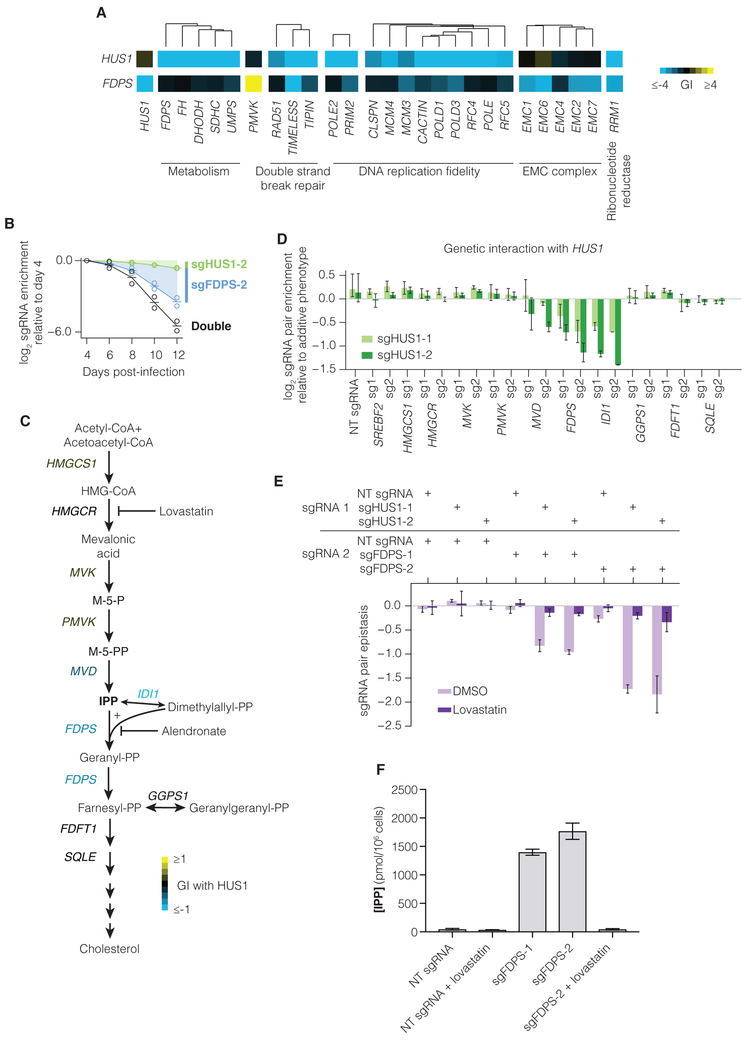
Seminal yeast studies have established the value of comprehensively mapping genetic interactions (GIs) for inferring gene function. Efforts in human cells using focused gene sets underscore the utility of this approach, but the feasibility of generating large-scale, diverse human GI maps remains unresolved. We developed a CRISPR interference platform for large-scale quantitative mapping of human GIs. We systematically perturbed 222,784 gene pairs in two cancer cell lines. The resultant maps cluster functionally related genes, assigning function to poorly characterized genes, including TMEM261, a new electron transport chain component. Individual GIs pinpoint unexpected relationships between pathways, exemplified by a specific cholesterol biosynthesis intermediate whose accumulation induces deoxynucleotide depletion, causing replicative DNA damage and a synthetic-lethal interaction with the ATR/9-1-1 DNA repair pathway. Our map provides a broad resource, establishes GI maps as a high-resolution tool for dissecting gene function, and serves as a blueprint for mapping the genetic landscape of human cells.
Keywords: CRISPR; CRISPRi; epistasis; functional genomics; genetic interactions.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures

References
-
- Baldwin KL, Dinh EM, Hart BM, and Masson PH (2013). CACTIN is an essential nuclear protein in Arabidopsis and may be associated with the eukaryotic spliceosome. FEBS Lett. 587, 873–879. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 CA217882/CA/NCI NIH HHS/United States
- R00 CA204602/CA/NCI NIH HHS/United States
- T32 GM007618/GM/NIGMS NIH HHS/United States
- DP1 DK113598/DK/NIDDK NIH HHS/United States
- R01 AR073017/AR/NIAMS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- DP2 GM119139/GM/NIGMS NIH HHS/United States
- P30 AR057235/AR/NIAMS NIH HHS/United States
- R00 AG047255/AG/NIA NIH HHS/United States
- K99 AG047255/AG/NIA NIH HHS/United States
- U01 CA168370/CA/NCI NIH HHS/United States
- K99 CA181494/CA/NCI NIH HHS/United States
- R00 CA181494/CA/NCI NIH HHS/United States
- R01 AG062359/AG/NIA NIH HHS/United States
- K99 CA204602/CA/NCI NIH HHS/United States
- R01 NS091902/NS/NINDS NIH HHS/United States
- P50 GM102706/GM/NIGMS NIH HHS/United States
- R01 DA036858/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
