

Attenuation of oxidative damage by targeting mitochondrial complex I in neonatal hypoxic-ischemic brain injury
- PMID: 30037775
- PMCID: PMC6389362
- DOI: 10.1016/j.freeradbiomed.2018.06.040
Attenuation of oxidative damage by targeting mitochondrial complex I in neonatal hypoxic-ischemic brain injury
Abstract
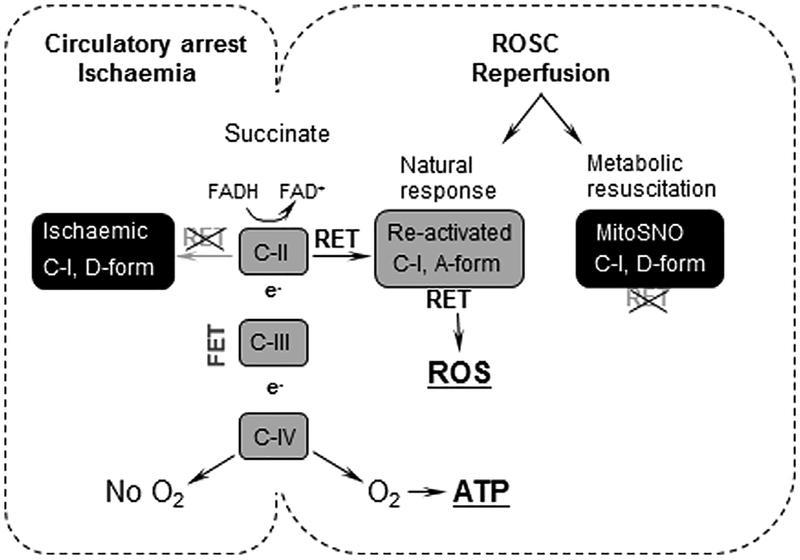
Background: Establishing sustained reoxygenation/reperfusion ensures not only the recovery, but may initiate a reperfusion injury in which oxidative stress plays a major role. This study offers the mechanism and this mechanism-specific therapeutic strategy against excessive release of reactive oxygen species (ROS) associated with reperfusion-driven recovery of mitochondrial metabolism.
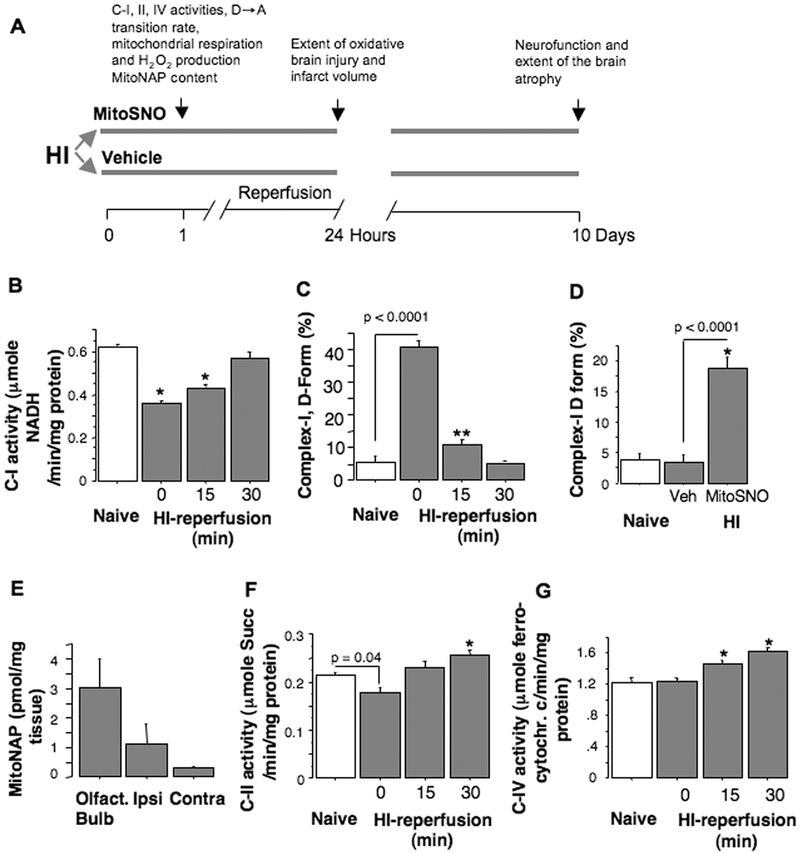
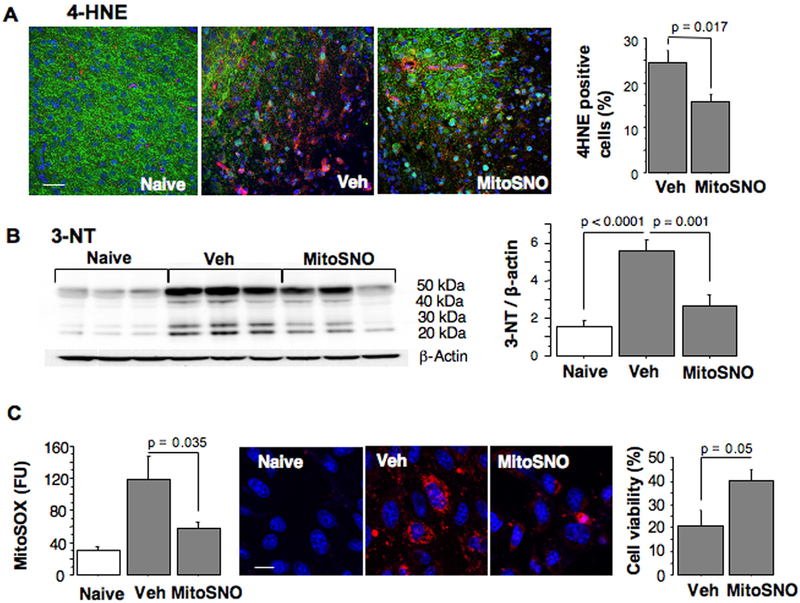
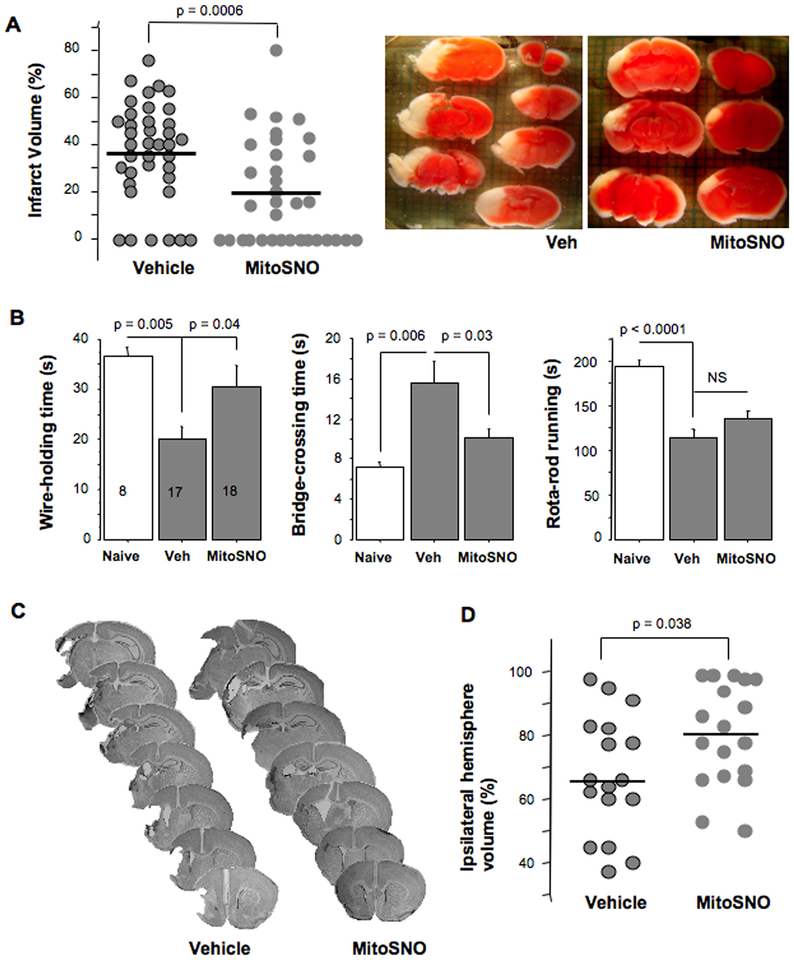
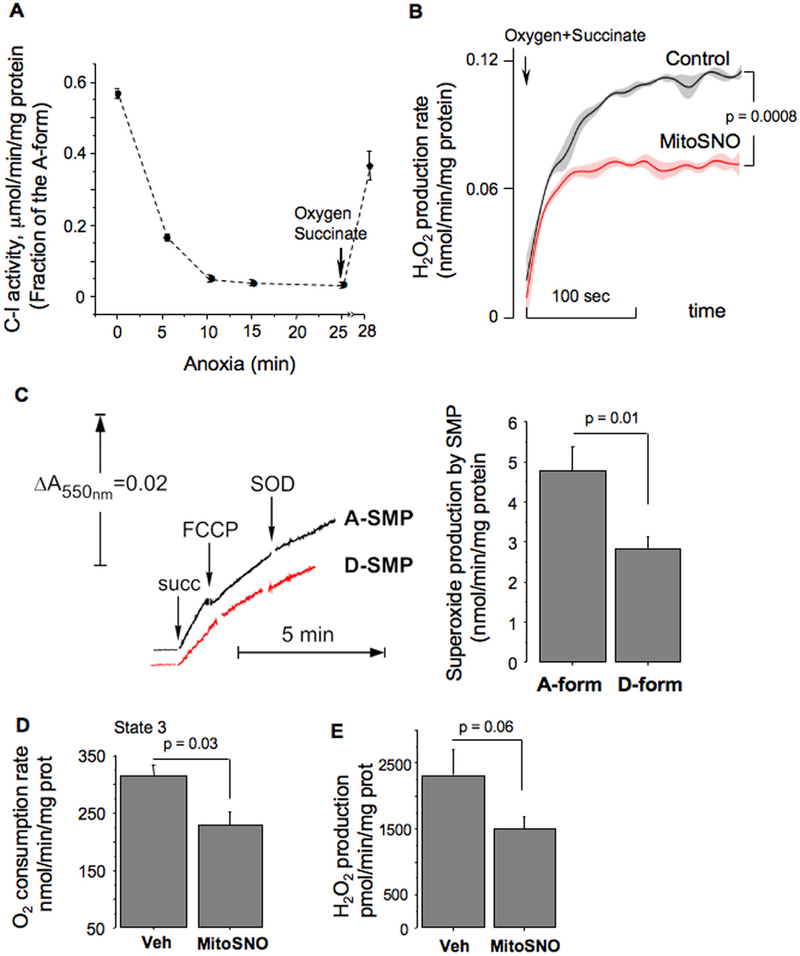
Aims and methods: In neonatal mice subjected to cerebral hypoxia-ischaemia (HI) and reperfusion, we examined conformational changes and activity of mitochondrial complex I with and without post-HI administration of S-nitrosating agent, MitoSNO. Assessment of mitochondrial ROS production, oxidative brain damage, neuropathological and neurofunctional outcomes were used to define neuroprotective strength of MitoSNO. A specificity of reperfusion-driven mitochondrial ROS production to conformational changes in complex I was examined in-vitro.
Results: HI deactivated complex I, changing its conformation from active form (A) into the catalytically dormant, de-active form (D). Reperfusion rapidly converted the D-form into the A-form and increased ROS generation. Administration of MitoSNO at the onset of reperfusion, decelerated D→A transition of complex I, attenuated oxidative stress, and significantly improved neurological recovery. In cultured neurons, after simulated ischaemia-reperfusion injury, MitoSNO significantly reduced ROS generation and neuronal mortality. In isolated mitochondria subjected to anoxia-reoxygenation, MitoSNO restricted ROS release during D→A transitions.
Conclusion: Rapid D→A conformation in response to reperfusion reactivates complex I. This is essential not only for metabolic recovery, but also contributes to excessive release of mitochondrial ROS and reperfusion injury. We propose that the initiation of reperfusion should be followed by pharmacologically-controlled gradual reactivation of complex I.
Keywords: Hypoxia/ischaemia; Ischaemia/reperfusion damage; Mitochondrial complex I; Nitrosation.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interest
None.
Figures
References
-
- Volpe JJ, Neonatal encephalopathy: an inadequate term for hypoxic-ischemic encephalopathy, Ann. Neurol 72 (2012) 156–166. - PubMed
-
- Piantadosi CA, Zhang J, Mitochondrial generation of reactive oxygen species after brain ischemia in the rat, Stroke 27 (1996) 327–331 (discussion 32). - PubMed
-
- Starkov AA, Chinopoulos C, Fiskum G, Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury, Cell Calcium 36 (2004) 257–264. - PubMed
-
- Ten VS, Starkov A, Hypoxic-ischemic injury in the developing brain: the role of reactive oxygen species originating in mitochondria, Neurol.Res.Int 2012 (2012) 542976. - PMC - PubMed
-
- Almeida A, Allen KL, Bates TE, Clark JB, Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain, J. Neurochem 65 (1995) 1698–1703. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
