

Perilipin 1 (Plin1) deficiency promotes inflammatory responses in lean adipose tissue through lipid dysregulation
- PMID: 30042231
- PMCID: PMC6130955
- DOI: 10.1074/jbc.RA118.003541
Perilipin 1 (Plin1) deficiency promotes inflammatory responses in lean adipose tissue through lipid dysregulation
Abstract
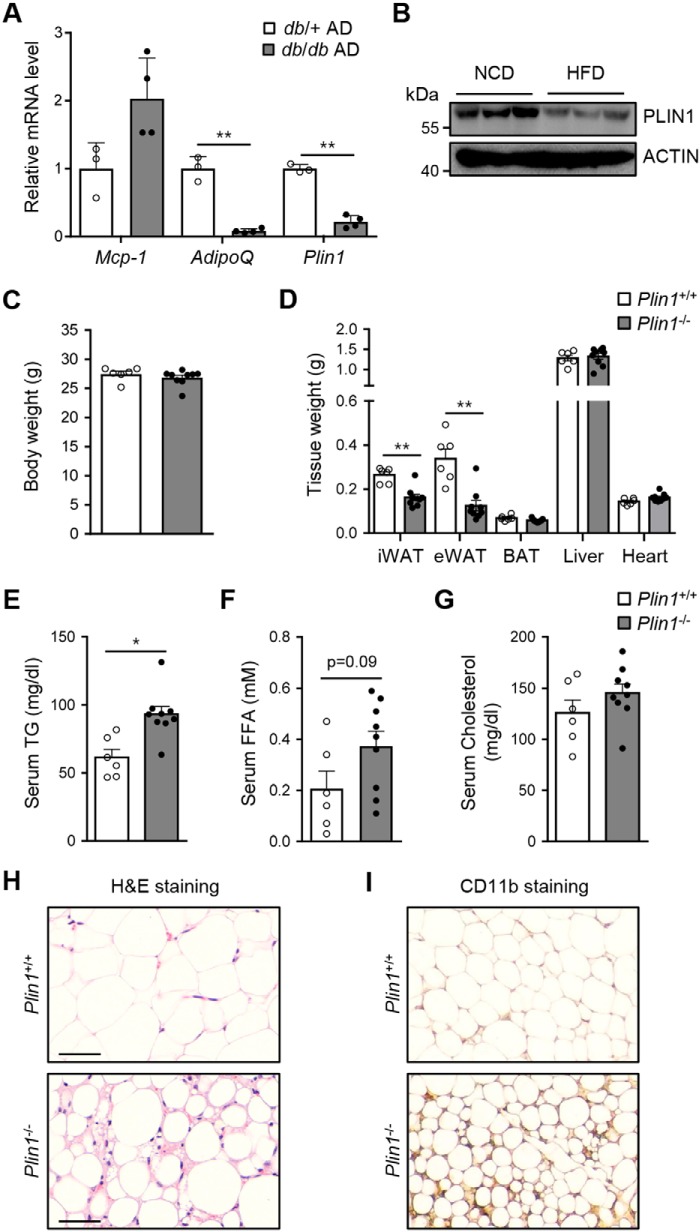
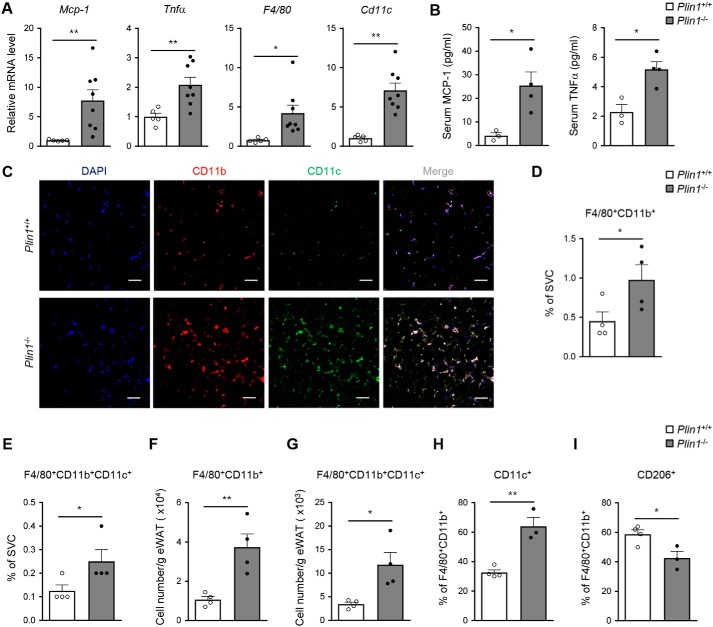
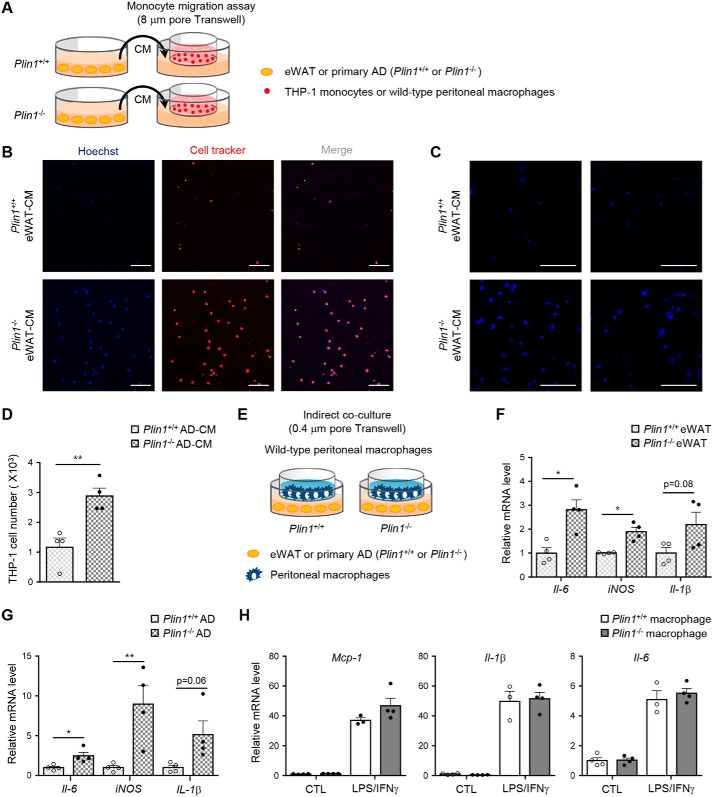
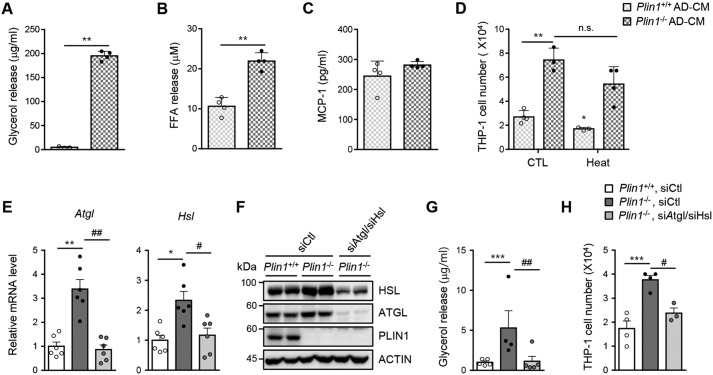
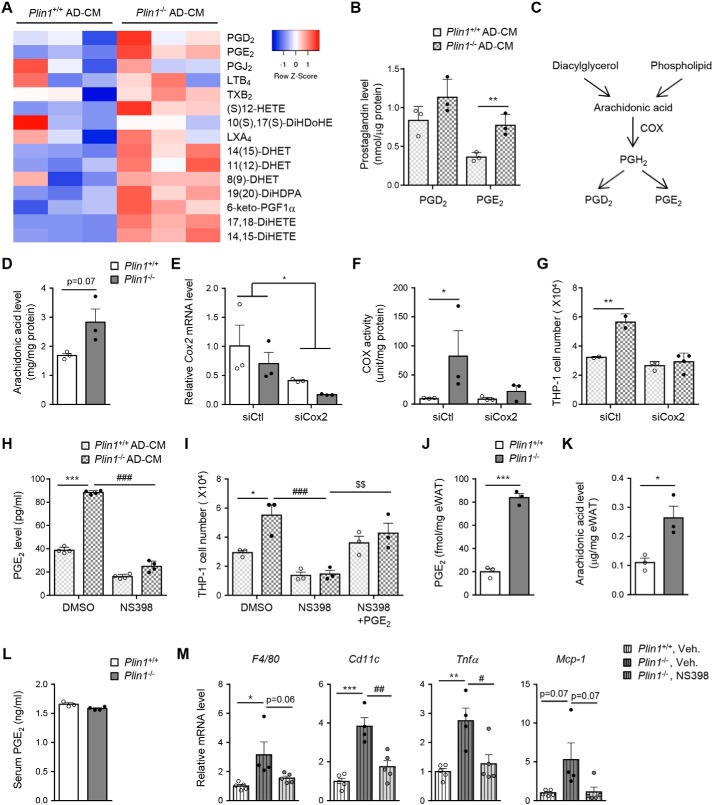
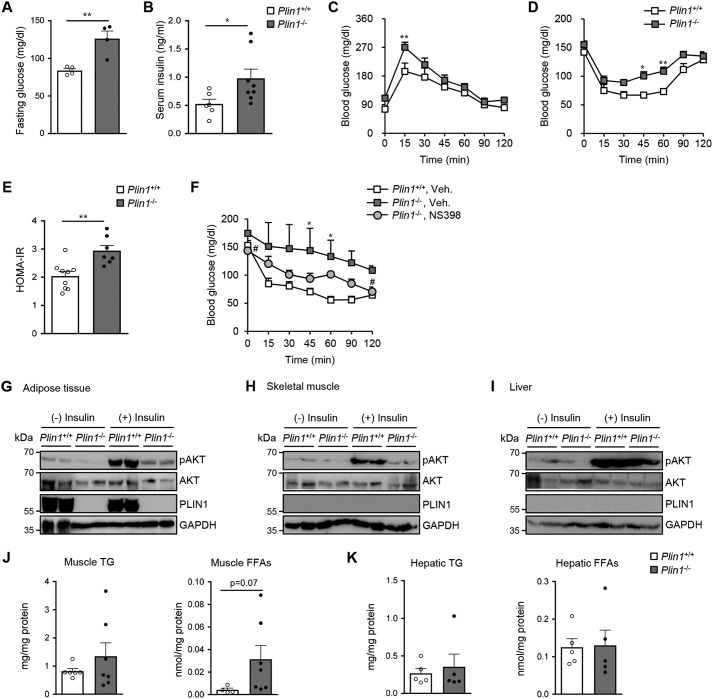
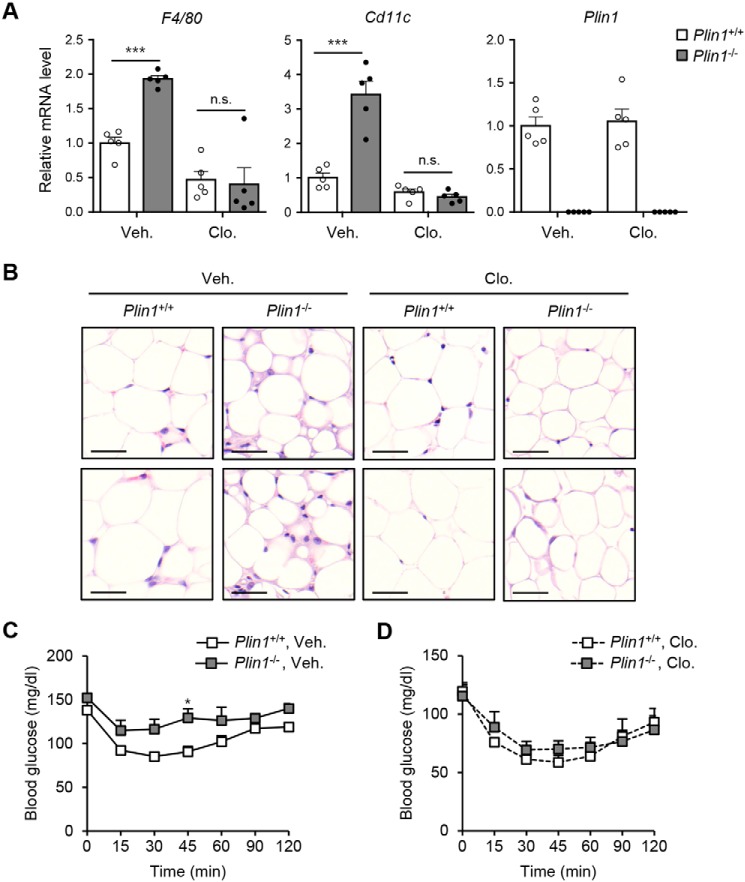
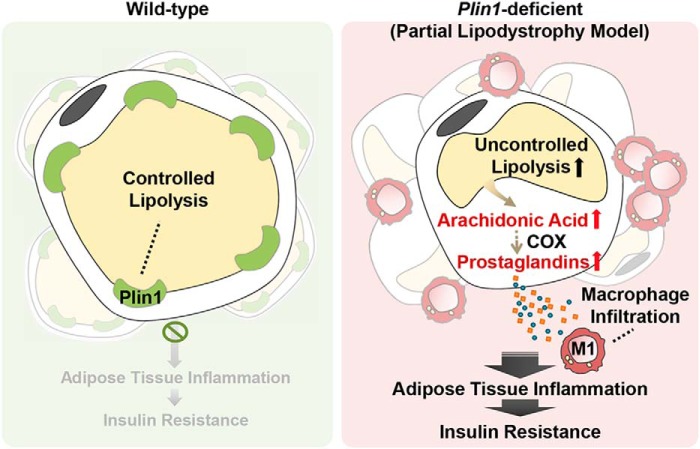
Lipid droplets are specialized cellular organelles that contain neutral lipid metabolites and play dynamic roles in energy homeostasis. Perilipin 1 (Plin1), one of the major lipid droplet-binding proteins, is highly expressed in adipocytes. In mice, Plin1 deficiency impairs peripheral insulin sensitivity, accompanied with reduced fat mass. However, the mechanisms underlying insulin resistance in lean Plin1 knockout (Plin1-/-) mice are largely unknown. The current study demonstrates that Plin1 deficiency promotes inflammatory responses and lipolysis in adipose tissue, resulting in insulin resistance. M1-type adipose tissue macrophages (ATMs) were higher in Plin1-/- than in Plin1+/+ mice on normal chow diet. Moreover, using lipidomics analysis, we discovered that Plin1-/- adipocytes promoted secretion of pro-inflammatory lipid metabolites such as prostaglandins, which potentiated monocyte migration. In lean Plin1-/- mice, insulin resistance was relieved by macrophage depletion with clodronate, implying that elevated pro-inflammatory ATMs might be attributable for insulin resistance under Plin1 deficiency. Together, these data suggest that Plin1 is required to restrain fat loss and pro-inflammatory responses in adipose tissue by reducing futile lipolysis to maintain metabolic homeostasis.
Keywords: Plin1; adipocyte; adipose tissue inflammation; adipose tissue macrophage; insulin resistance; lipid metabolism; lipolysis; prostaglandin.
© 2018 Sohn et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Xu H., Barnes G. T., Yang Q., Tan G., Yang D., Chou C. J., Sole J., Nichols A., Ross J. S., Tartaglia L. A., and Chen H. (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830 10.1172/JCI200319451 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources