

Mitochondrial quality control mechanisms as molecular targets in cardiac ageing
- PMID: 30042431
- PMCID: PMC6283278
- DOI: 10.1038/s41569-018-0059-z
Mitochondrial quality control mechanisms as molecular targets in cardiac ageing
Abstract
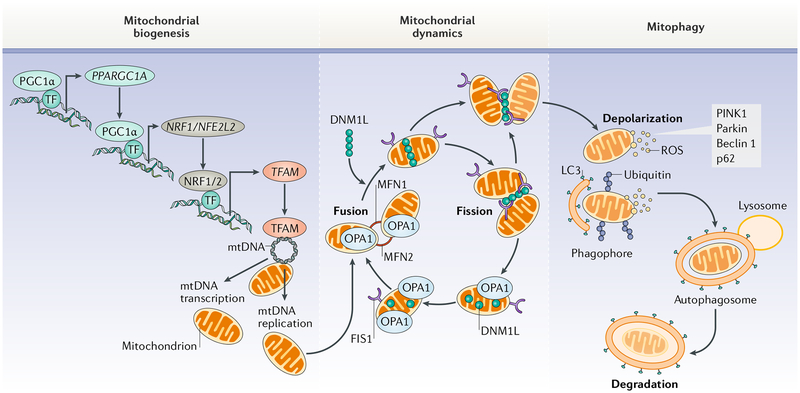
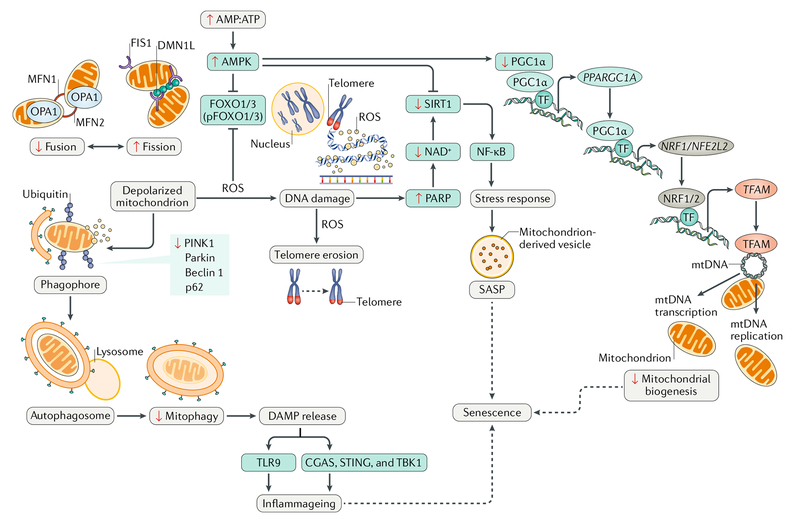
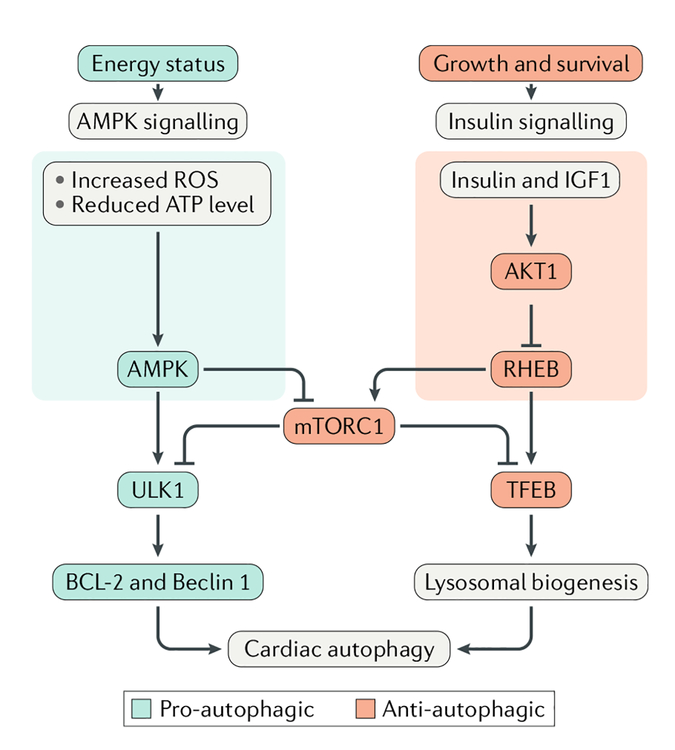
Cardiovascular disease is the leading cause of morbidity and mortality worldwide. Advancing age is a major risk factor for developing cardiovascular disease because of the lifelong exposure to cardiovascular risk factors and specific alterations affecting the heart and the vasculature during ageing. Indeed, the ageing heart is characterized by structural and functional changes that are caused by alterations in fundamental cardiomyocyte functions. In particular, the myocardium is heavily dependent on mitochondrial oxidative metabolism and is especially susceptible to mitochondrial dysfunction. Indeed, primary alterations in mitochondrial function, which are subsequently amplified by defective quality control mechanisms, are considered to be major contributing factors to cardiac senescence. In this Review, we discuss the mechanisms linking defective mitochondrial quality control mechanisms (that is, proteostasis, biogenesis, dynamics, and autophagy) to organelle dysfunction in the context of cardiac ageing. We also illustrate relevant molecular pathways that might be exploited for the prevention and treatment of age-related heart dysfunction.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Mozaffarian D et al. Heart disease and stroke statistics—2016 update. Circulation 133, e38–e360 (2016). - PubMed
-
- Baris OR et al. Mosaic deficiency in mitochondrial oxidative metabolism promotes cardiac arrhythmia during aging. Cell Metab. 21, 667–677 (2015). - PubMed
-
- Lok NS & Lau CP Prevalence of palpitations, cardiac arrhythmias and their associated risk factors in ambulant elderly. Int. J. Cardiol 54, 231–236 (1996). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
