

Overriding Traditional Electronic Effects in Biocatalytic Baeyer-Villiger Reactions by Directed Evolution
- PMID: 30044629
- PMCID: PMC6314816
- DOI: 10.1021/jacs.8b04742
Overriding Traditional Electronic Effects in Biocatalytic Baeyer-Villiger Reactions by Directed Evolution
Abstract
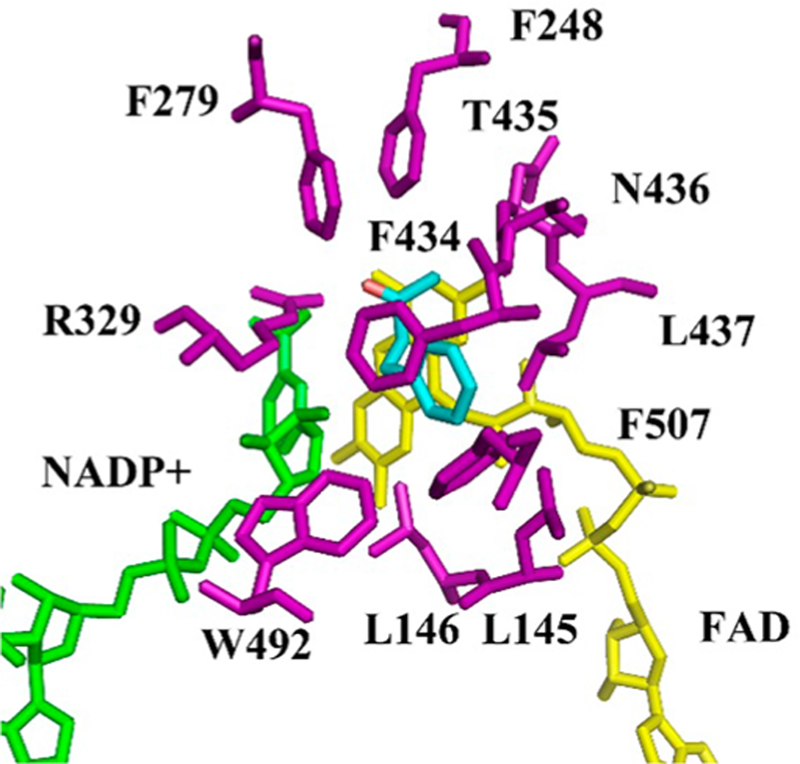
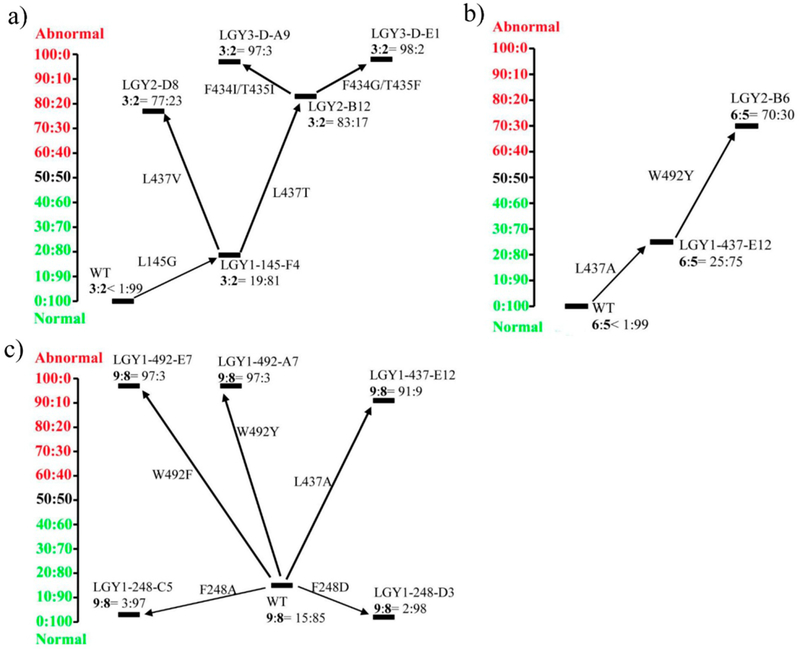
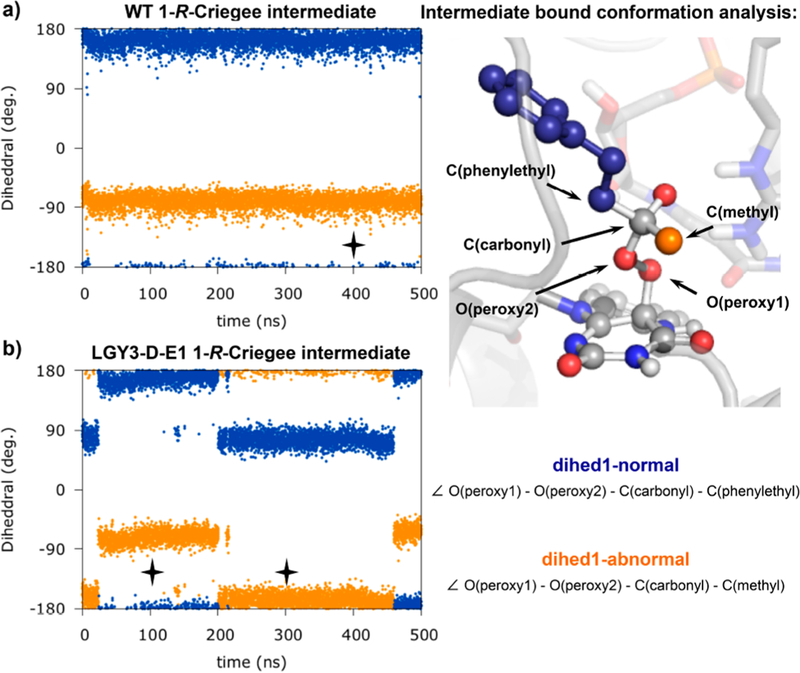
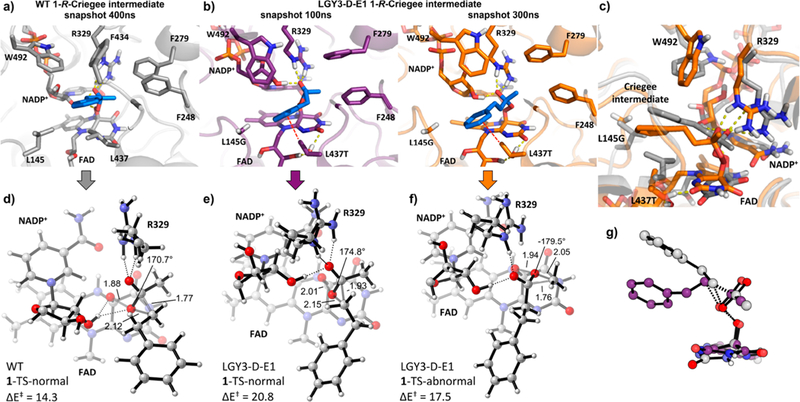
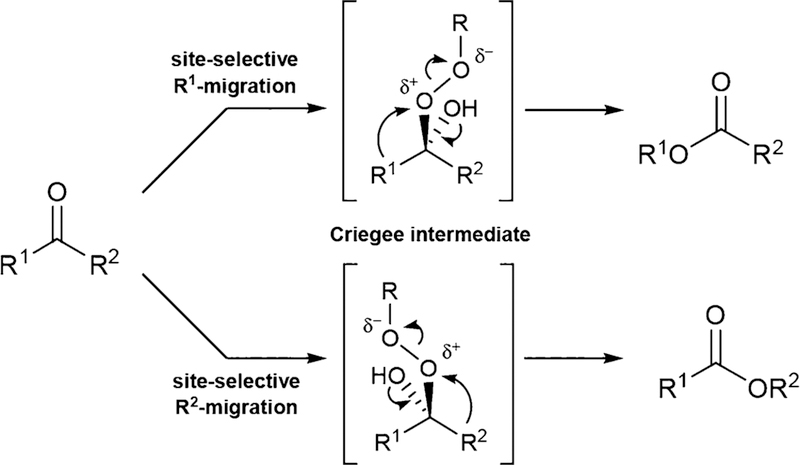
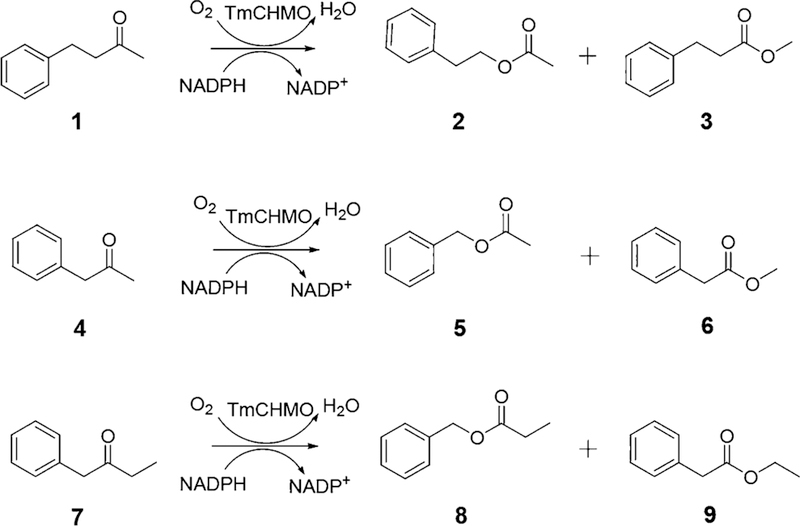
Controlling the regioselectivity of Baeyer-Villiger (BV) reactions remains an ongoing issue in organic chemistry, be it by synthetic catalysts or enzymes of the type Baeyer-Villiger monooxygenases (BVMOs). Herein, we address the challenging problem of switching normal to abnormal BVMO regioselectivity by directed evolution using three linear ketones as substrates, which are not structurally biased toward abnormal reactivity. Upon applying iterative saturation mutagenesis at sites lining the binding pocket of the thermostable BVMO from Thermocrispum municipale DSM 44069 (TmCHMO) and using 4-phenyl-2-butanone as substrate, the regioselectivity was reversed from 99:1 (wild-type enzyme in favor of the normal product undergoing 2-phenylethyl migration) to 2:98 in favor of methyl migration when applying the best mutant. This also stands in stark contrast to the respective reaction using the synthetic reagent m-CPBA, which provides solely the normal product. Reversal of regioselectivity was also achieved in the BV reaction of two other linear ketones. Kinetic parameters and melting temperatures revealed that most of the evolved mutants retained catalytic activity, as well as thermostability. In order to shed light on the origin of switched regioselectivity in reactions of 4-phenyl-2-butanone and phenylacetone, extensive QM/MM and MD simulations were performed. It was found that the mutations introduced by directed evolution induce crucial changes in the conformation of the respective Criegee intermediates and transition states in the binding pocket of the enzyme. In mutants that destabilize the normally preferred migration transition state, a reversal of regioselectivity is observed. This conformational control of regioselectivity overrides electronic control, which normally causes preferential migration of the group that is best able to stabilize positive charge. The results can be expected to aid future protein engineering of BVMOs.
Conflict of interest statement
Notes
The authors declare no competing financial interest.
Figures
References
-
- Baeyer A; Villiger V Ber. Dtsch. Chem. Ges 1899, 32, 3625–3633.
-
- Reviews of Baeyer−Villiger reactions: (a) Krow GR Org. React 1993, 43, 251–256.
- (b) Renz M; Meunier B Eur. J. Org. Chem 1999, 4, 737–750.
- (c) Ten Brink G-J; Arends IWCE; Sheldon RA Chem. Rev 2004, 104, 4105–4124. - PubMed
- (d) Mihovilovic MD; Rudroff F; Grötzl B Curr. Org. Chem 2004, 8, 1057–1069.
- (e) Jiménez-Sanchidriań C; Ruiz JR Tetrahedron 2008, 64, 2011–2026.
- (f) Uyanik M; Ishihara K ACS Catal 2013, 3, 513–520.
-
- Asymmetric BV reactions using synthetic transition metal catalysts: (a) Bolm C; Schlingloff G; Weickhardt K Angew. Chem., Int. Ed. Engl 1994, 33, 1848–1849.
- (b) Ito K; Ishii A; Kuroda T; Katsuki T Synlett 2003, 643–646.
- (c) Strukul G Angew. Chem., Int. Ed 1998, 37, 1198–1209. - PubMed
- (d) Frison J-C; Palazzi C; Bolm C Tetrahedron 2006, 62, 6700–6706.
- (e) Malkov AV; Friscourt F; Bell M; Swarbrick ME; Kocovsky P J. Org. Chem 2008, 73, 3996–4003. - PubMed
- (f) Zhou L; Liu X; Ji J; Zhang Y; Hu X; Lin L; Feng XJ Am. Chem. Soc 2012, 134, 17023–17026. - PubMed
- (g) Zhou L; Liu X; Ji J; Zhang Y; Wu W; Liu Y; Lin L; Feng X Org. Lett 2014, 16, 3938–3941. - PubMed
- (h) Poudel PP; Arimitsu K; Yamamoto K Chem. Commun 2016, 52, 4163–4166. - PubMed
- (i) Russo A; De Fusco C; Lattanzi A ChemCatChem 2012, 4, 901–916.
-
- Asymmetric BV reactions using chiral organocatalysts: (a) Murahashi S; Ono S; Imada Y Angew. Chem., Int. Ed 2002, 41, 2366–2370. - PubMed
- (b) Imada Y; Iida H; Murahashi SI; Naota T Angew. Chem., Int. Ed 2005, 44, 1704–1706. - PubMed
- (c) Wang B; Shen YM; Shi YJ Org. Chem 2006, 71, 9519–9521. - PubMed
- (d) Peris G; Miller SJ Org. Lett 2008, 10, 3049–3052. - PMC - PubMed
- (e) Xu S; Wang Z; Zhang X; Ding K Angew. Chem., Int. Ed 2008, 47, 2840–2843. - PubMed
- (f) Abascal NC; Miller SJ Org. Lett 2016, 18, 4646–4649. - PMC - PubMed
-
- (a) Yadav VK Steric and Stereoelectronic Effects in Organic Chemistry; Springer, 2016.
- (b) Alabugin IV Stereoelectronic Effects: A Bridge between Structure and Reactivity; John Wiley & Sons, 2016.
- (c) Deslongchamps P Stereoelectronic Effects in Organic Chemistry; Pergamon Press: New York, 1983.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
