

Intercellular Spread of Protein Aggregates in Neurodegenerative Disease
- PMID: 30044648
- PMCID: PMC6350082
- DOI: 10.1146/annurev-cellbio-100617-062636
Intercellular Spread of Protein Aggregates in Neurodegenerative Disease
Abstract
Most neurodegenerative diseases are characterized by the accumulation of protein aggregates, some of which are toxic to cells. Mounting evidence demonstrates that in several diseases, protein aggregates can pass from neuron to neuron along connected networks, although the role of this spreading phenomenon in disease pathogenesis is not completely understood. Here we briefly review the molecular and histopathological features of protein aggregation in neurodegenerative disease, we summarize the evidence for release of proteins from donor cells into the extracellular space, and we highlight some other mechanisms by which protein aggregates might be transmitted to recipient cells. We also discuss the evidence that supports a role for spreading of protein aggregates in neurodegenerative disease pathogenesis and some limitations of this model. Finally, we consider potential therapeutic strategies to target spreading of protein aggregates in the treatment of neurodegenerative diseases.
Keywords: neurodegenerative disease; protein aggregate; seeding; spreading.
Figures
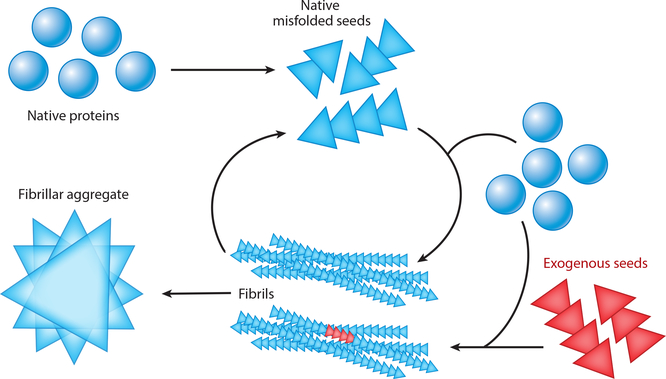
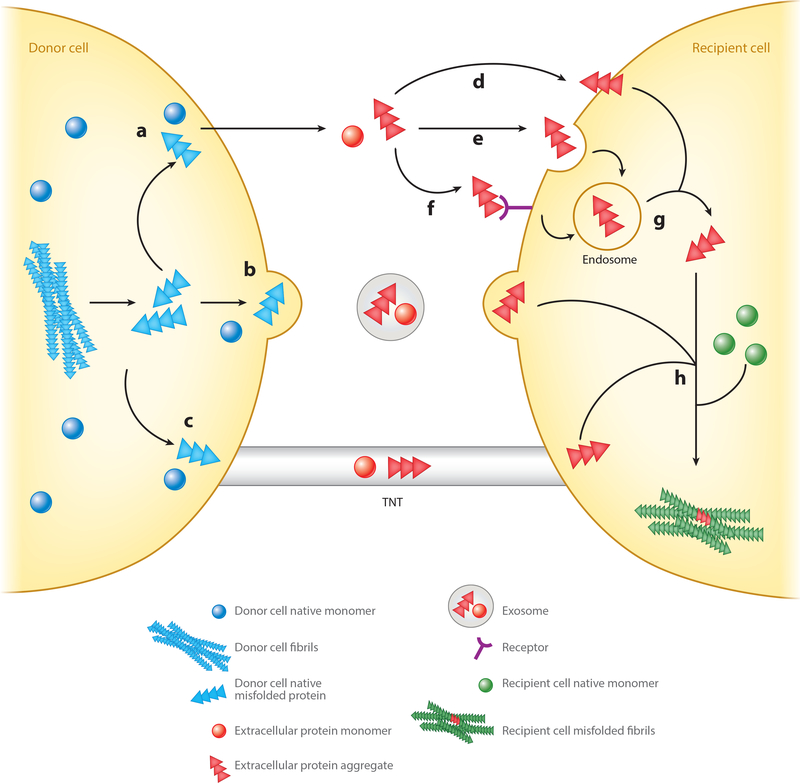
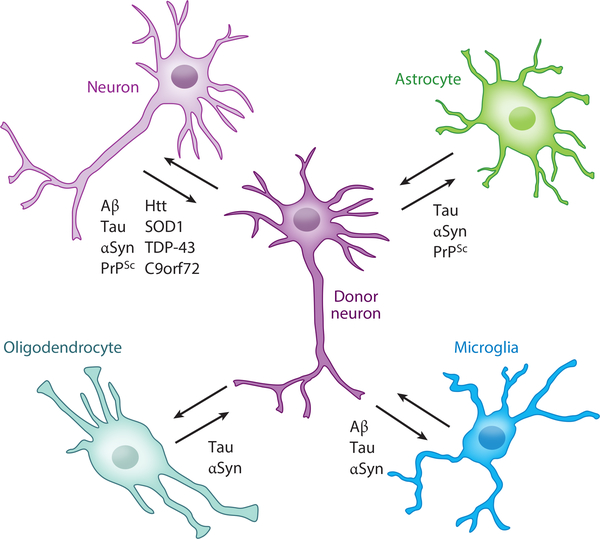
References
-
- Affiris AG. 2014. Studies assessing tolerability and safety of AFFITOPE® PD03A in patients with early Parkinson’s disease (AFF011). Rep. NCT02267434, US Natl. Libr. Med., Natl. Inst. Health, Washington, DC: https://clinicaltrials.gov/ct2/show/NCT02267434
-
- Alzheimer A 1907. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr 64:146–48
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
