

The RUNX1/IL-34/CSF-1R axis is an autocrinally regulated modulator of resistance to BRAF-V600E inhibition in melanoma
- PMID: 30046005
- PMCID: PMC6124424
- DOI: 10.1172/jci.insight.120422
The RUNX1/IL-34/CSF-1R axis is an autocrinally regulated modulator of resistance to BRAF-V600E inhibition in melanoma
Abstract
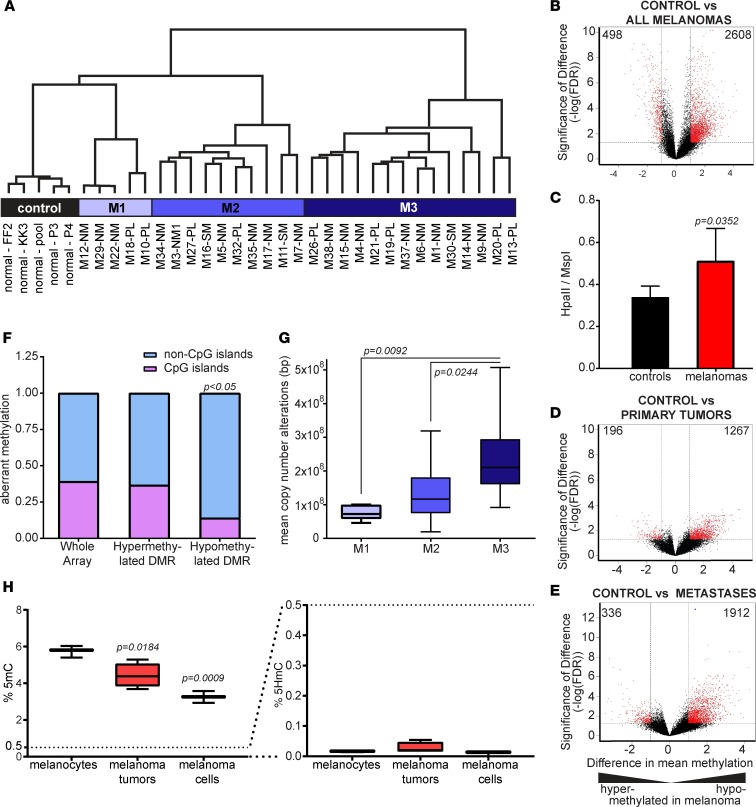
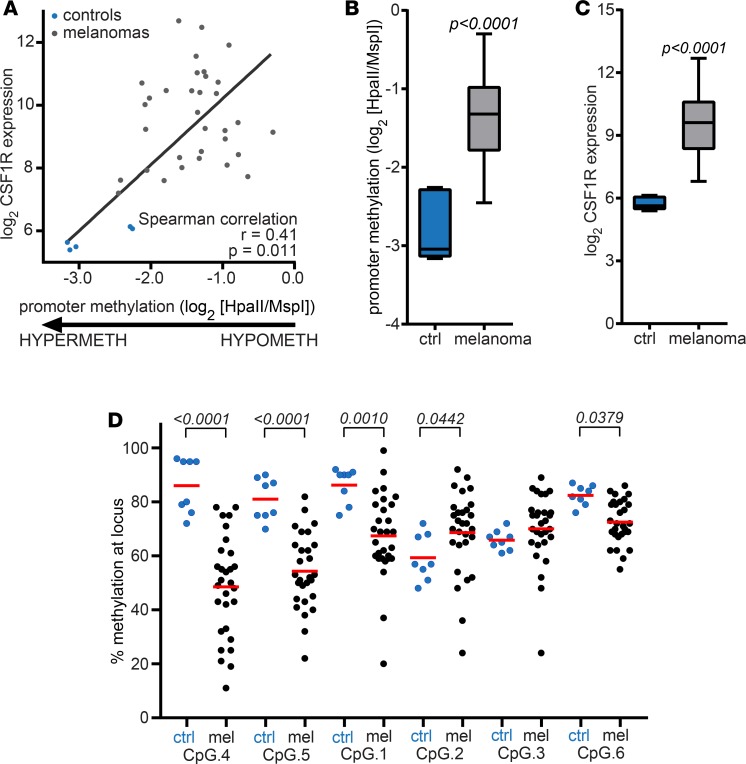
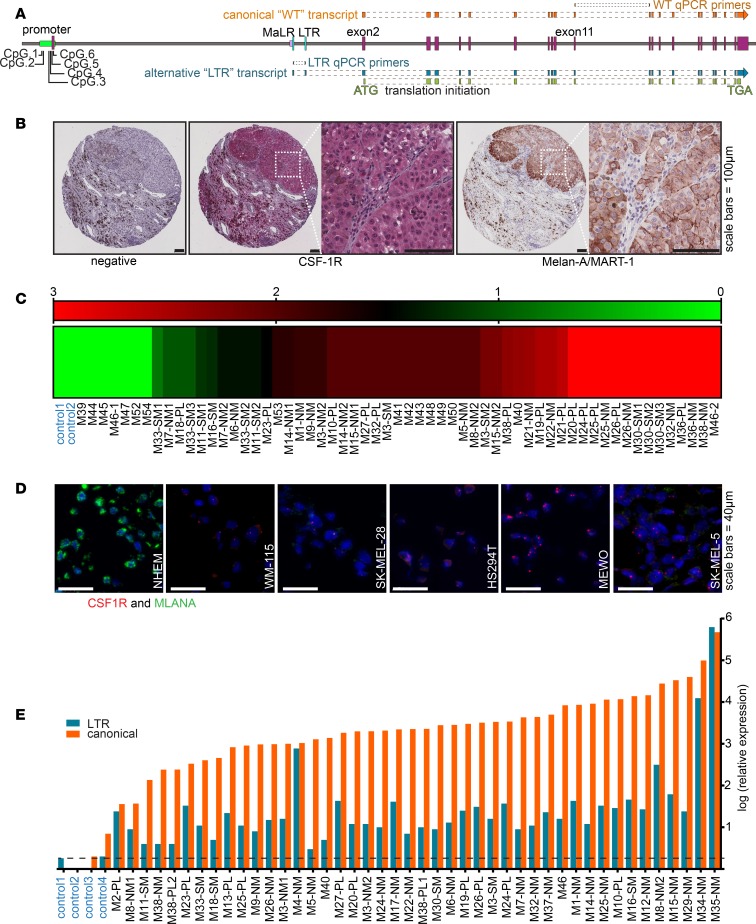
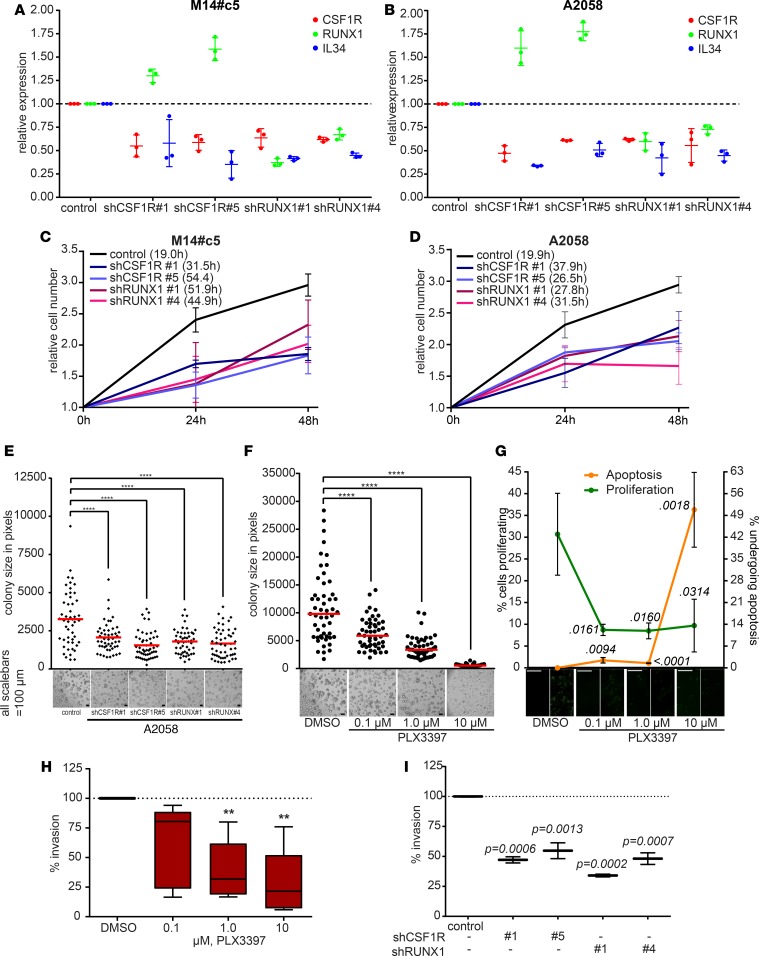
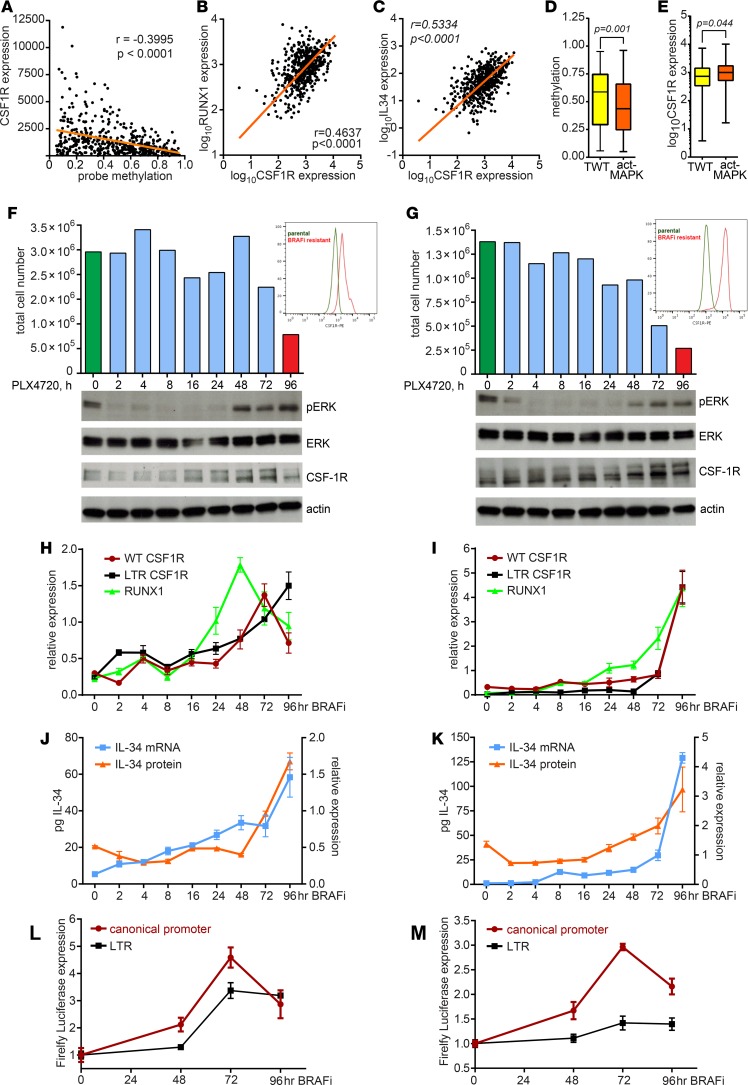
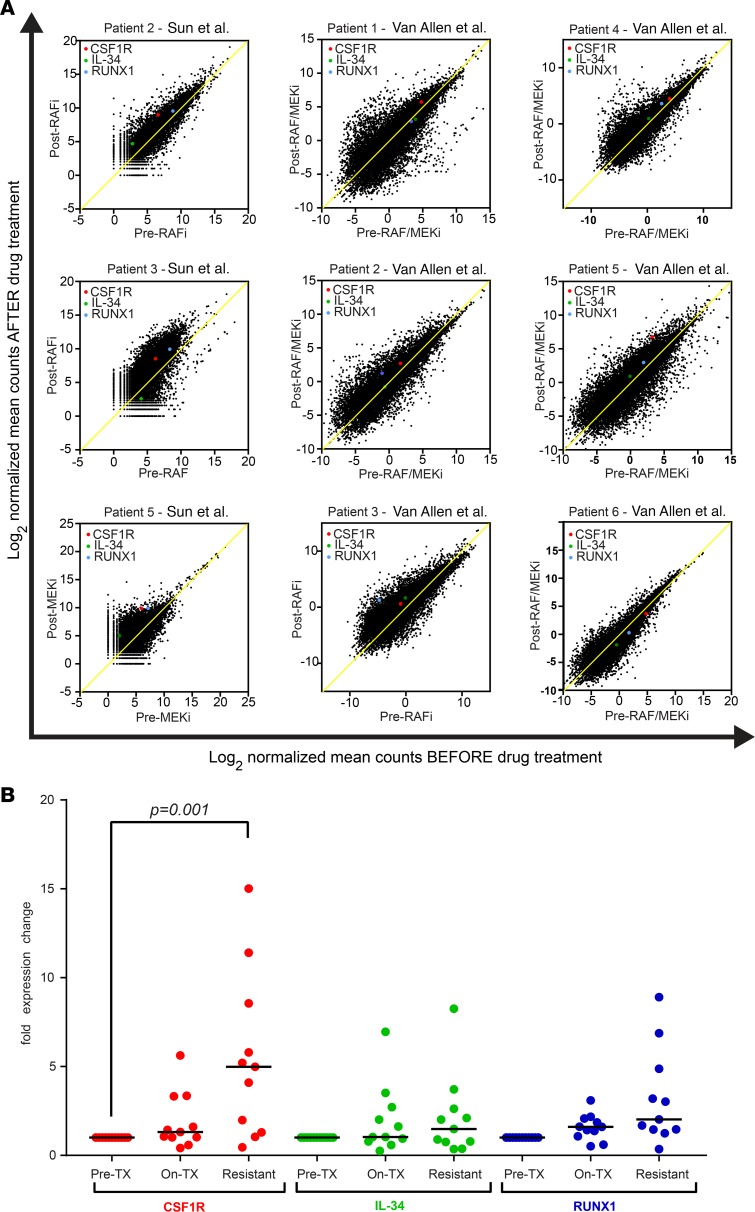
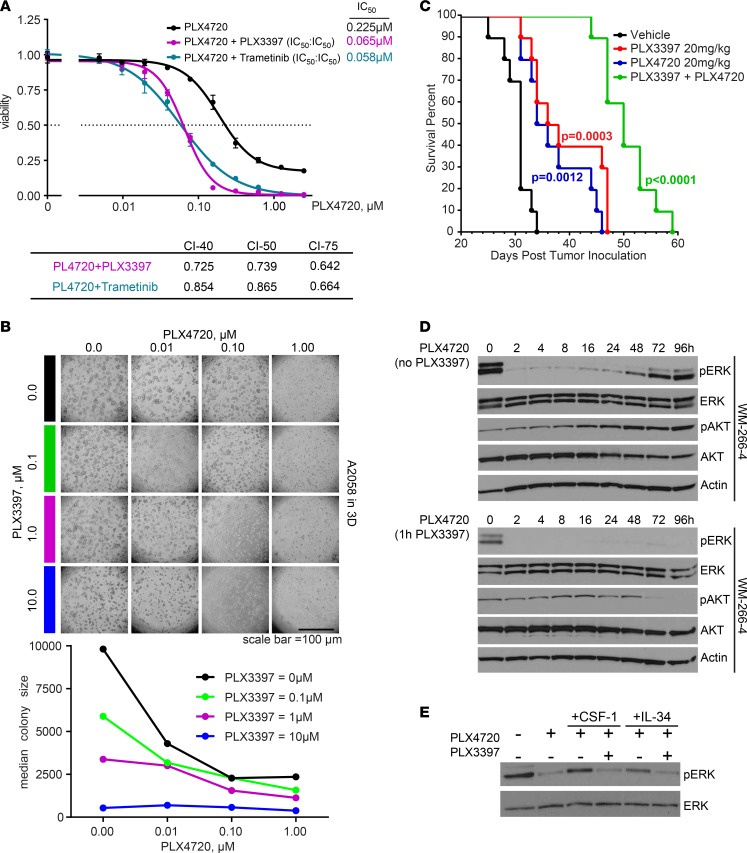
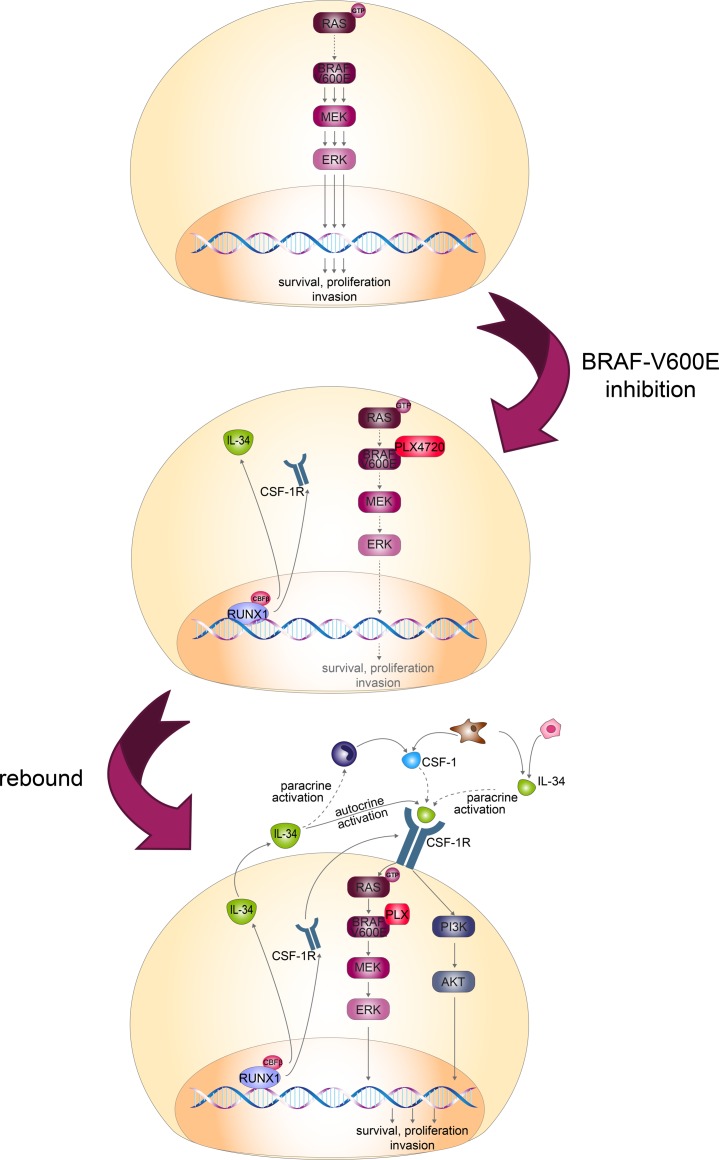
Resistance to current therapies still impacts a significant number of melanoma patients and can be regulated by epigenetic alterations. Analysis of global cytosine methylation in a cohort of primary melanomas revealed a pattern of early demethylation associated with overexpression of oncogenic transcripts. Loss of methylation and associated overexpression of the CSF 1 receptor (CSF1R) was seen in a majority of tumors and was driven by an alternative, endogenous viral promoter in a subset of samples. CSF1R was particularly elevated in melanomas with BRAF and other MAPK activating mutations. Furthermore, rebound ERK activation after BRAF inhibition was associated with RUNX1-mediated further upregulation of CSF-1R and its ligand IL-34. Importantly, increased CSF-1R and IL-34 overexpression were detected in an independent cohort of resistant melanomas. Inhibition of CSF-1R kinase or decreased CSF-1R expression by RNAi reduced 3-D growth and invasiveness of melanoma cells. Coinhibition of CSF-1R and BRAF resulted in synergistic efficacy in vivo. To our knowledge, our data unveil a previously unknown role for the autocrine-regulated CSF-1R in BRAF V600E resistance and provide a preclinical rationale for targeting this pathway in melanoma.
Keywords: Melanoma; Oncology.
Conflict of interest statement
Figures
References
-
- Howlader N, et al. SEER Cancer Statistics Review, (CSR) 1975-2015. National Cancer Institute. https://seer.cancer.gov/csr/1975_2015/ Published April 16, 2018. Accessed June 27, 2018.
-
- Wolchok JD, et al. Updated results from a phase III trial of nivolumab (NIVO) combined with ipilimumab (IPI) in treatment-naive patients (pts) with advanced melanoma (MEL) (CheckMate 067) J Clin Oncol. 2016;34(15_suppl):9505
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
