

Cyclic nucleotide-dependent inhibitory signaling interweaves with activating pathways to determine platelet responses
- PMID: 30046761
- PMCID: PMC6046581
- DOI: 10.1002/rth2.12122
Cyclic nucleotide-dependent inhibitory signaling interweaves with activating pathways to determine platelet responses
Abstract
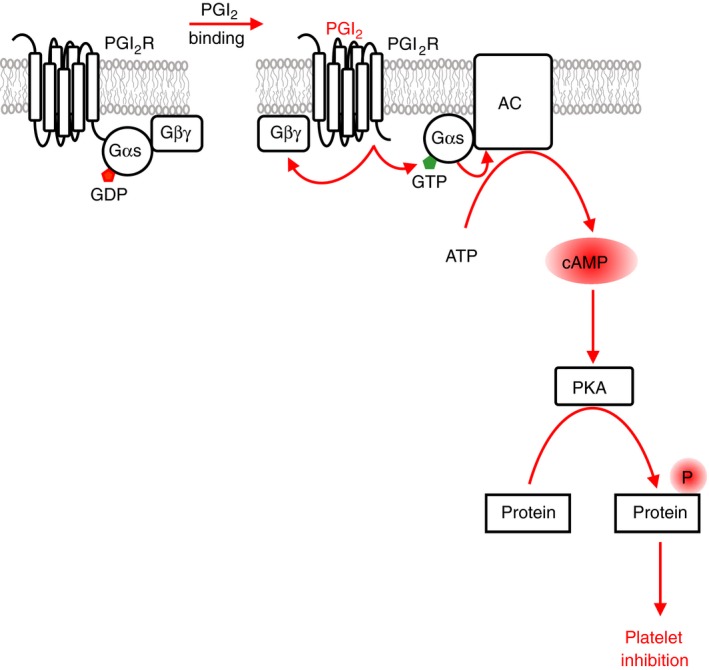
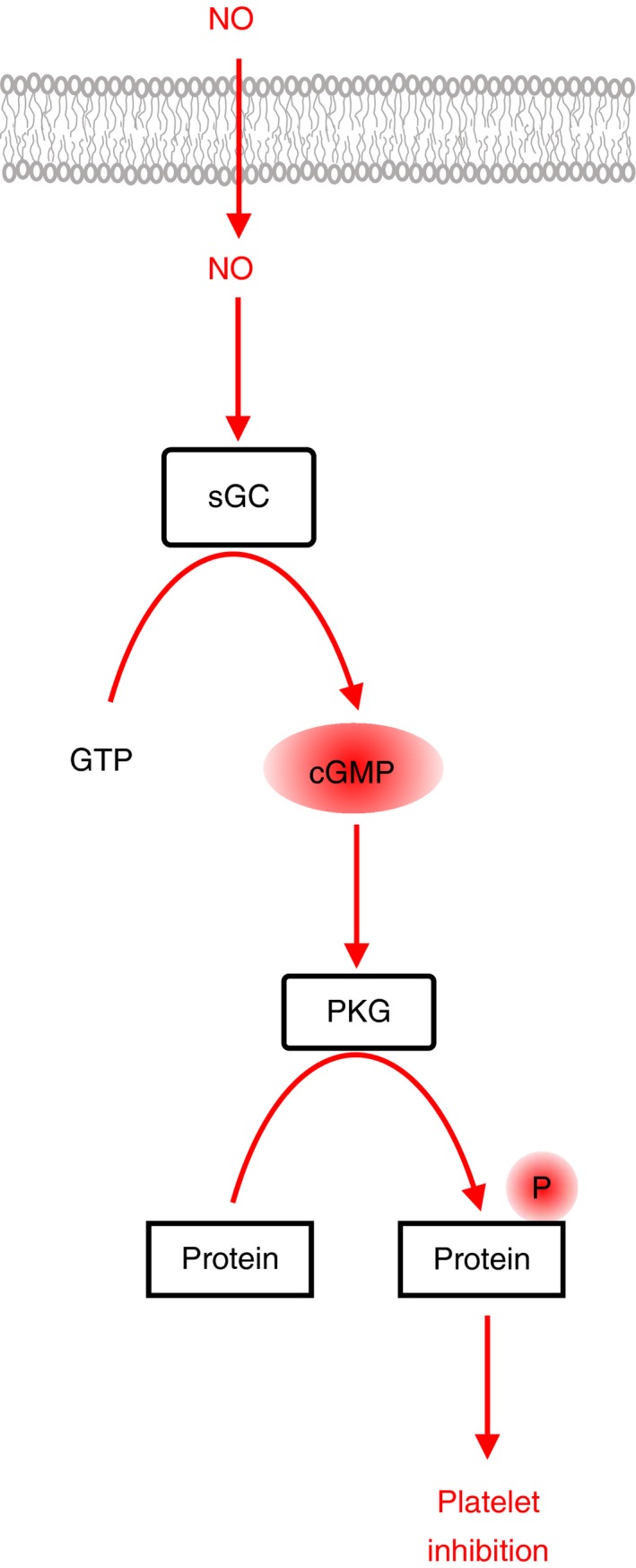
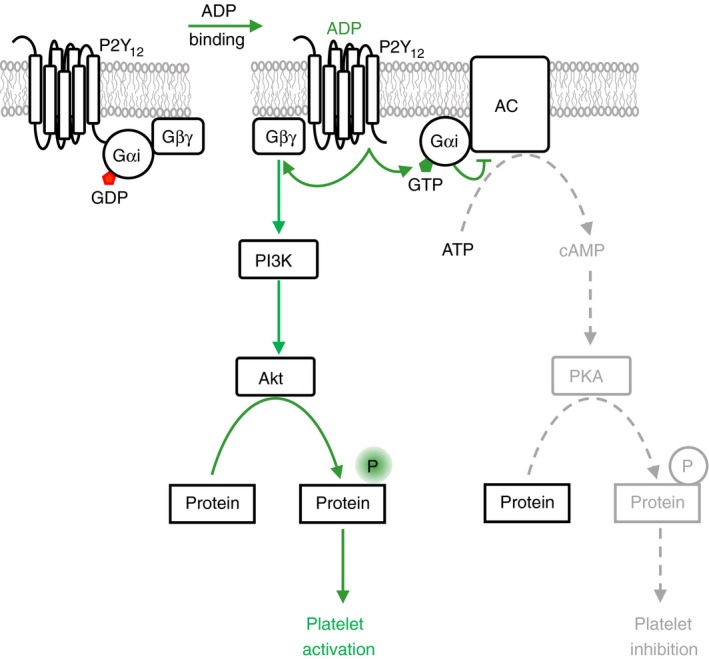
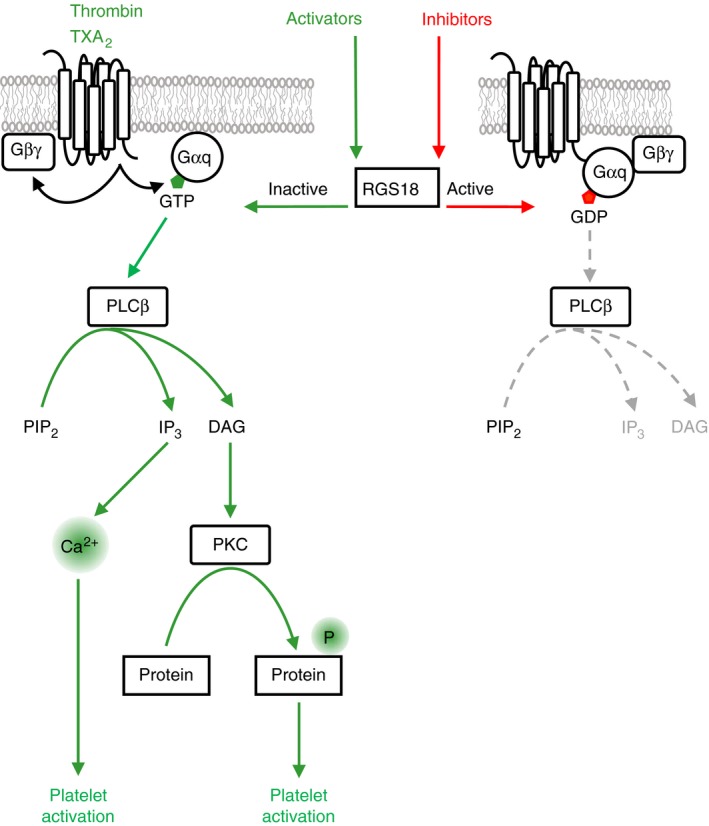
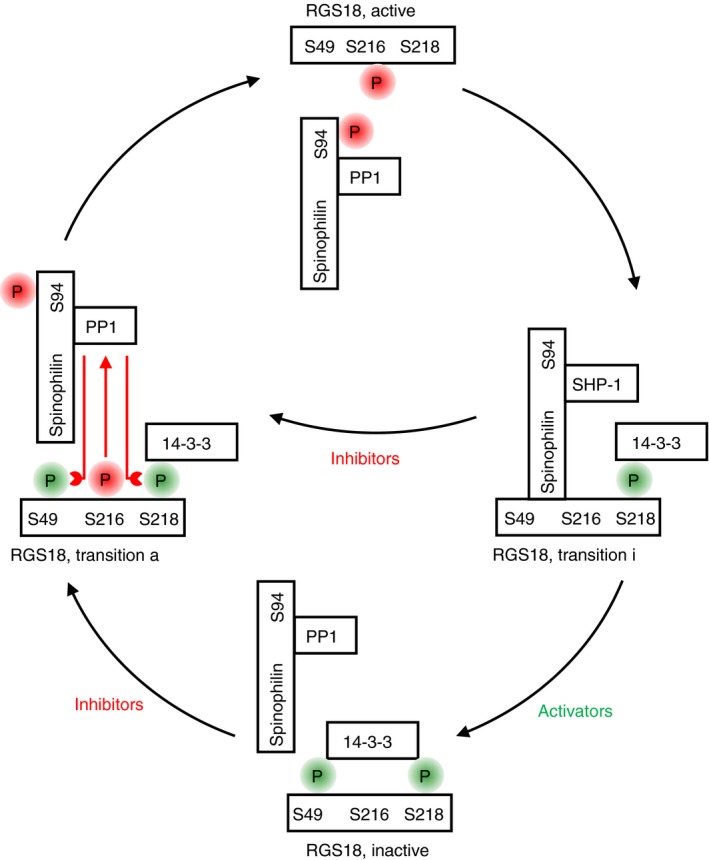
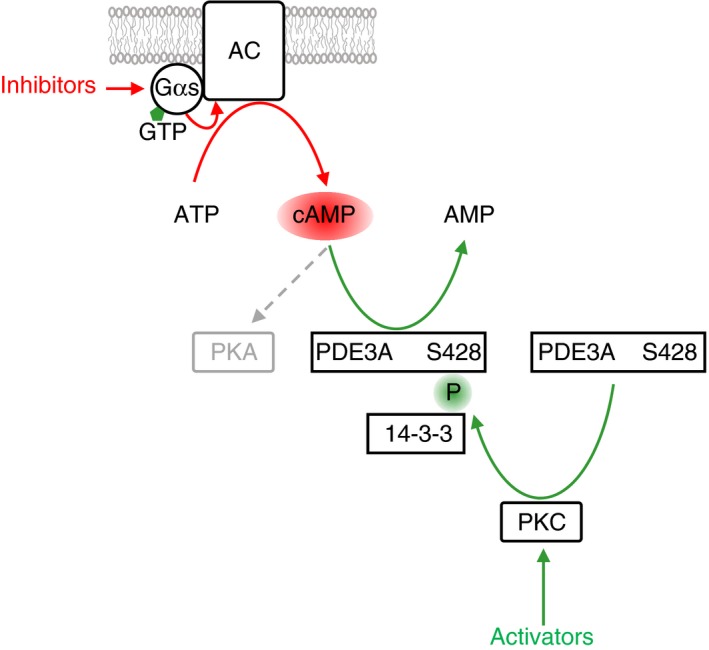
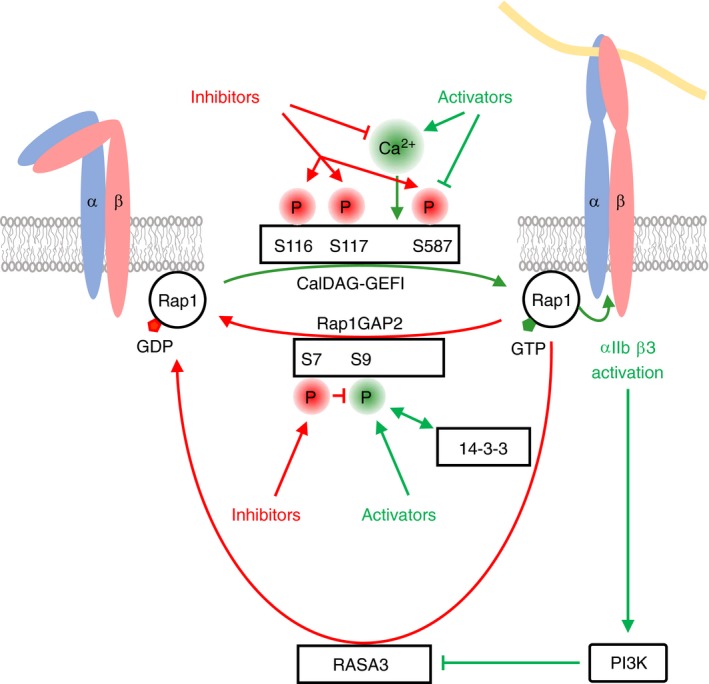
Platelets are regulated by extracellular cues that impact on intracellular signaling. The endothelium releases prostacyclin and nitric oxide which stimulate the synthesis of cyclic nucleotides cAMP and cGMP leading to platelet inhibition. Other inhibitory mechanisms involve immunoreceptor tyrosine-based inhibition motif-containing receptors, intracellular receptors and receptor desensitization. Inhibitory cyclic nucleotide pathways are traditionally thought to represent a passive background system keeping platelets in a quiescent state. In contrast, cyclic nucleotides are increasingly seen to be dynamically involved in most aspects of platelet regulation. This review focuses on crosstalk between activating and cyclic nucleotide-mediated inhibitory pathways highlighting emerging new hub structures and signaling mechanisms. In particular, interactions of plasma membrane receptors like P2Y12 and GPIb/IX/V with the cyclic nucleotide system are described. Furthermore, differential regulation of the RGS18 complex, second messengers, protein kinases, and phosphatases are presented, and control over small G-proteins by guanine-nucleotide exchange factors and GTPase-activating proteins are outlined. Possible clinical implications of signaling crosstalk are discussed.
Keywords: 14‐3‐3 protein; G‐protein; RGS18; cyclic AMP; kinase; protein kinase A.
Figures
References
-
- Smolenski A. Novel roles of cAMP/cGMP‐dependent signaling in platelets. J Thromb Haemost. 2012;10:167–76. - PubMed
-
- Walter U, Gambaryan S. cGMP and cGMP‐dependent protein kinase in platelets and blood cells. Handb Exp Pharmacol. 2009;191:533–48. - PubMed
-
- Clemetson KJ. Platelets and primary haemostasis. Thromb Res. 2012;129:220–4. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
