

Avoiding diagnostic delay for mucopolysaccharidosis IIIB: do not overlook common clues such as wheezing and otitis media
- PMID: 30049674
- PMCID: PMC6067142
- DOI: 10.1136/bcr-2018-224412
Avoiding diagnostic delay for mucopolysaccharidosis IIIB: do not overlook common clues such as wheezing and otitis media
Abstract
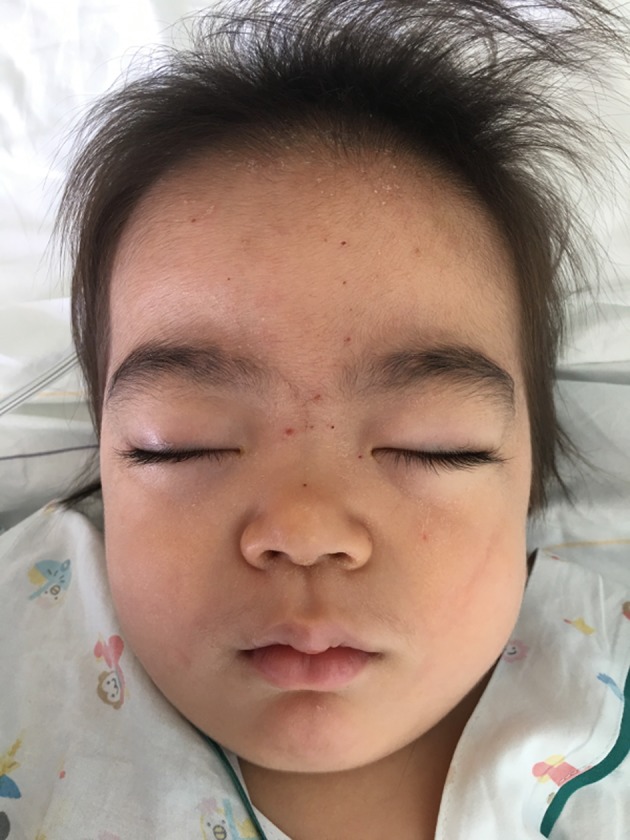
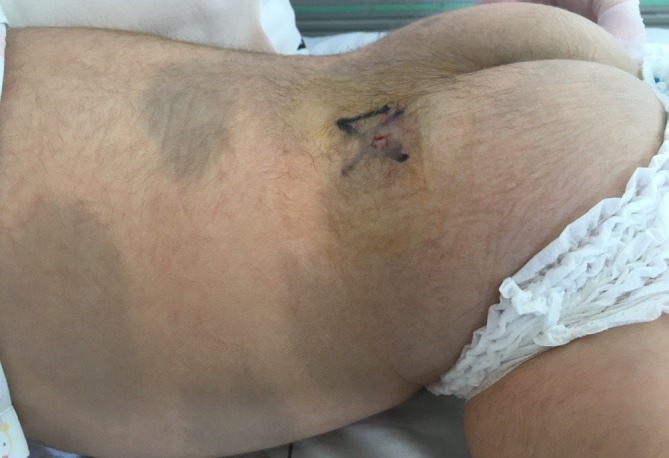
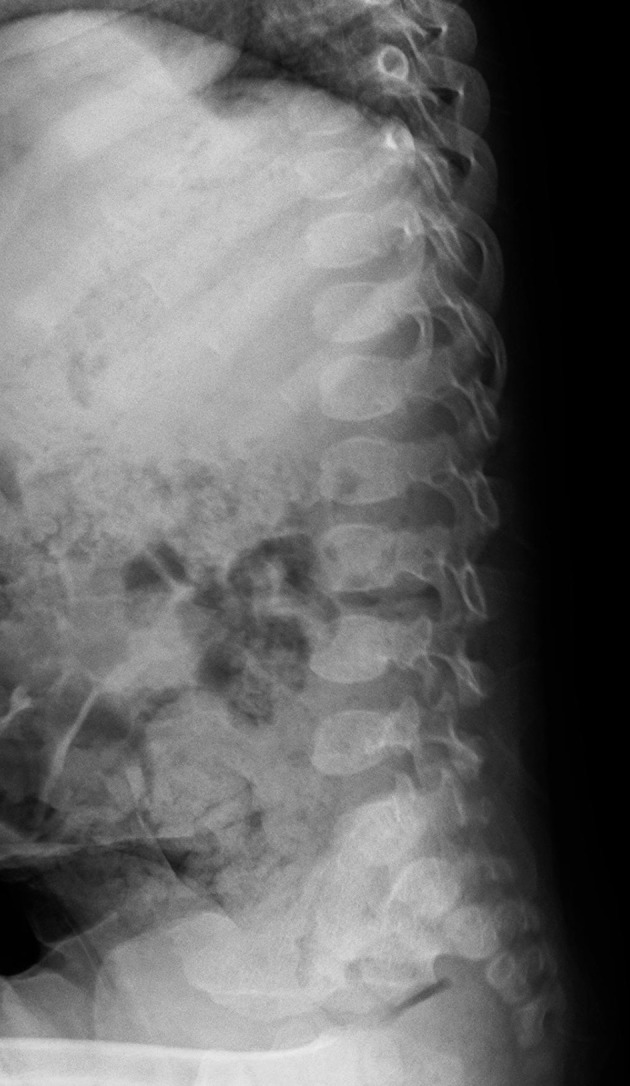
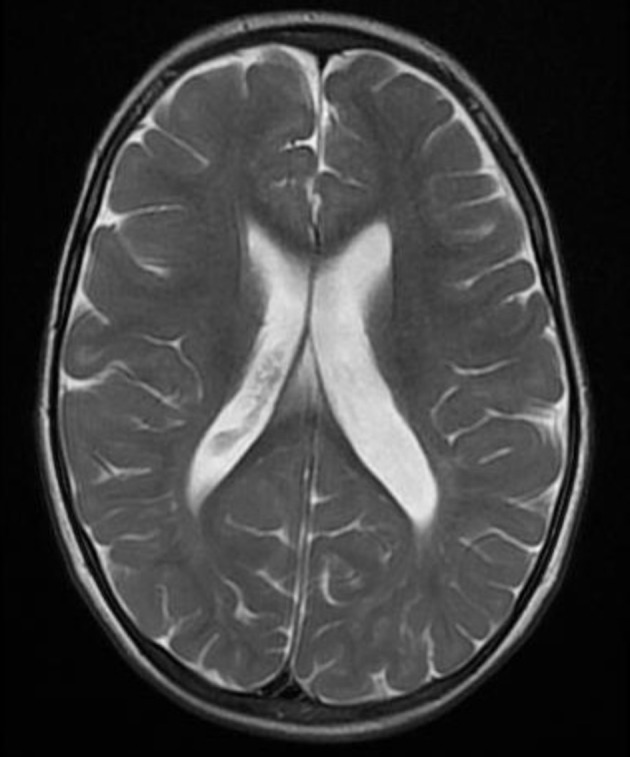
Mucopolysaccharidosis IIIB (MPS IIIB) is an autosomal recessive lysosomal storage disorder. In comparison to Hurler syndrome (MPS I) and Hunter syndrome (MPS II), characteristic facial and physical features tend to be milder and progression of neurological symptoms may initially be slower. Obvious neurological and behavioural symptoms may not appear until age 2-6 years, but once they begin, progression is relentless, leading to death by the early 20s. Although there is currently no known cure for MPS IIIB, enzyme replacement clinical trials are showing hope for delay in the progression of symptoms. Early diagnosis is therefore necessary before neurological symptoms have progressed. In our case, MPS IIIB was diagnosed at an early age because recurrent wheezing and otitis media in conjunction with hepatomegaly were recognised as more than trivial findings. A thorough examination and a definitive proactive decision to perform a liver biopsy resulted in early diagnosis of a rare disease.
Keywords: asthma; congenital disorders; genetics; liver disease; paediatrics.
© BMJ Publishing Group Limited 2018. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
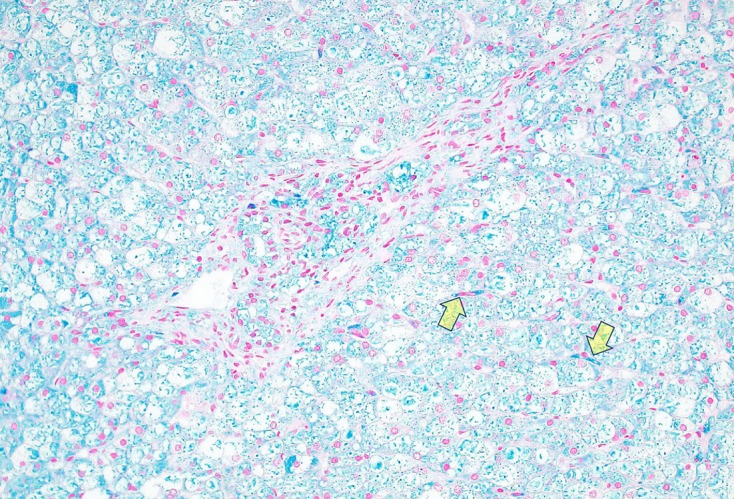
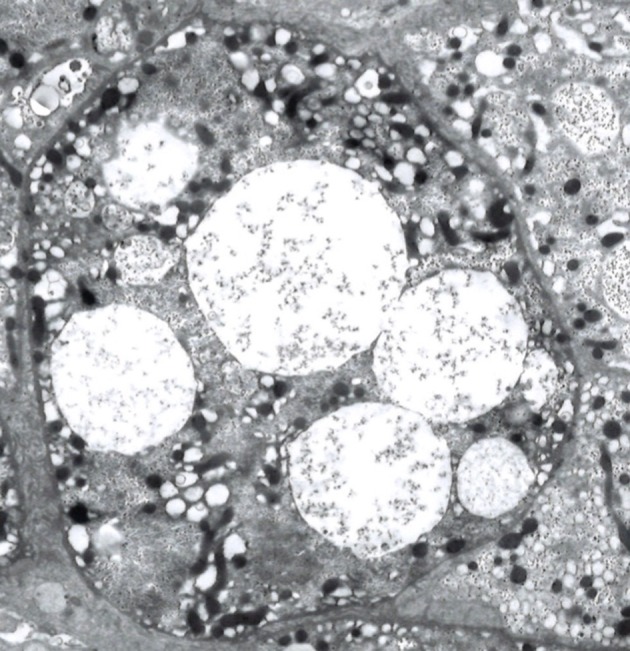
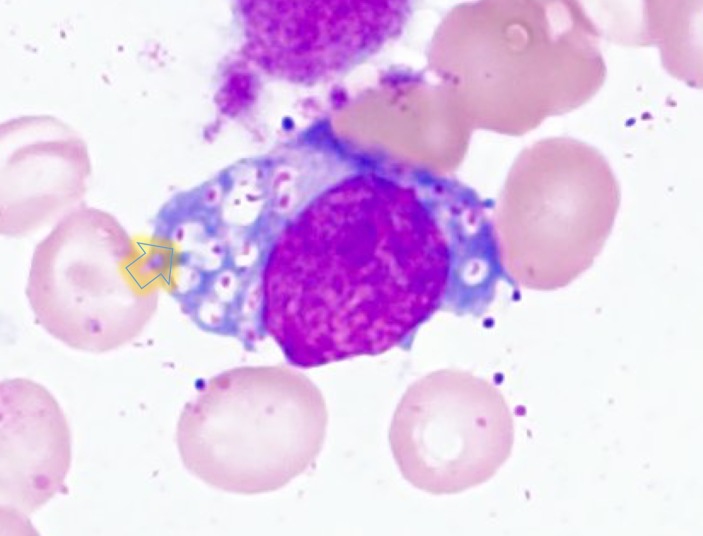
References
-
- Sanfilippo SJ, Podosin R, Langer L, et al. . Mental retardation associated with acid mucopolysacchariduria (heparitin sulfate type). J Pediatr 1963;63:837–8. 10.1016/S0022-3476(63)80279-6 - DOI
-
- Tanaka A, Kimura M, Lan HT, et al. . Molecular analysis of the alpha-N-acetylglucosaminidase gene in seven Japanese patients from six unrelated families with mucopolysaccharidosis IIIB (Sanfilippo type B), including two novel mutations. J Hum Genet 2002;47:484–7. 10.1007/s100380200070 - DOI - PubMed
-
- Chuang SC, Hwu WL, Wu CC, et al. . Diagnosis of mucopolysaccharidosis type IIIB. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi 1996;37:320–3. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources