

Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts
- PMID: 30052023
- PMCID: PMC6763210
- DOI: 10.1021/acs.chemrev.7b00678
Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts
Abstract
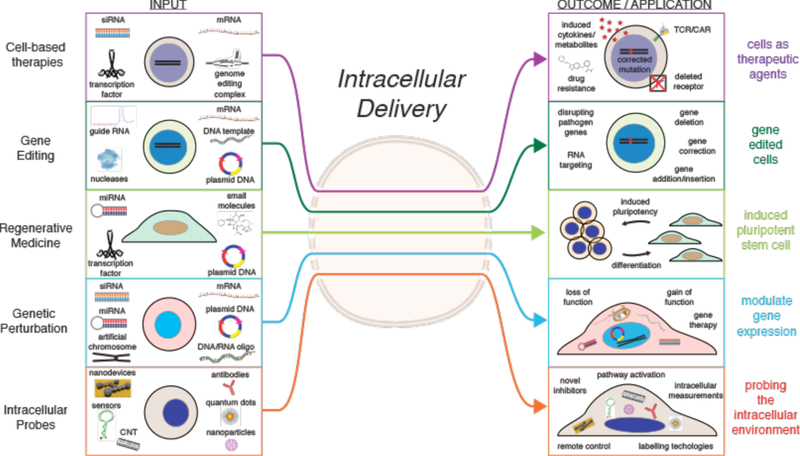
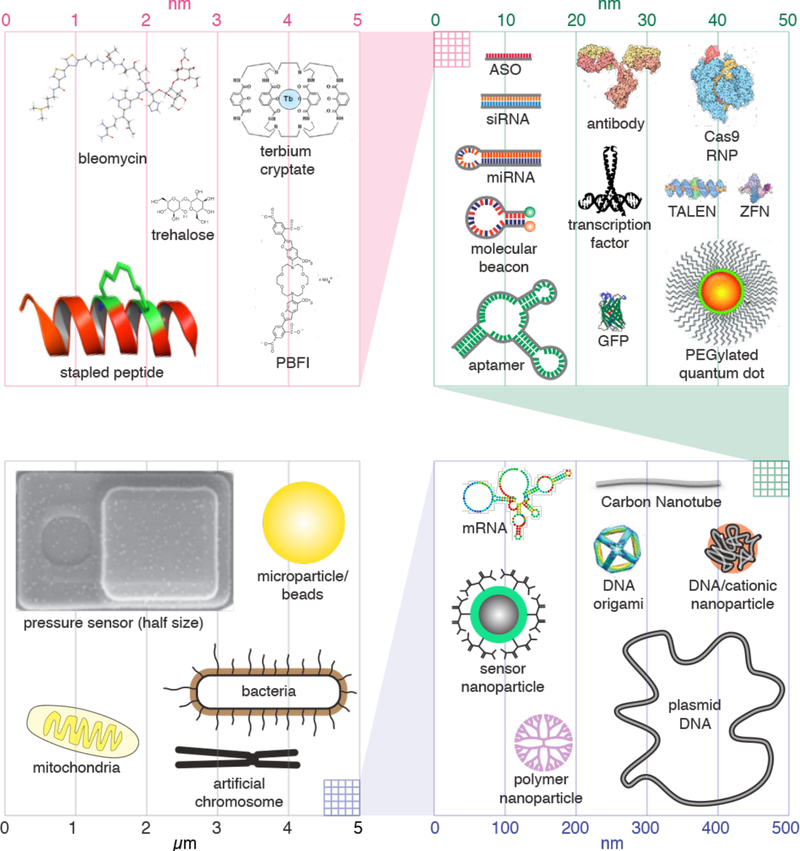
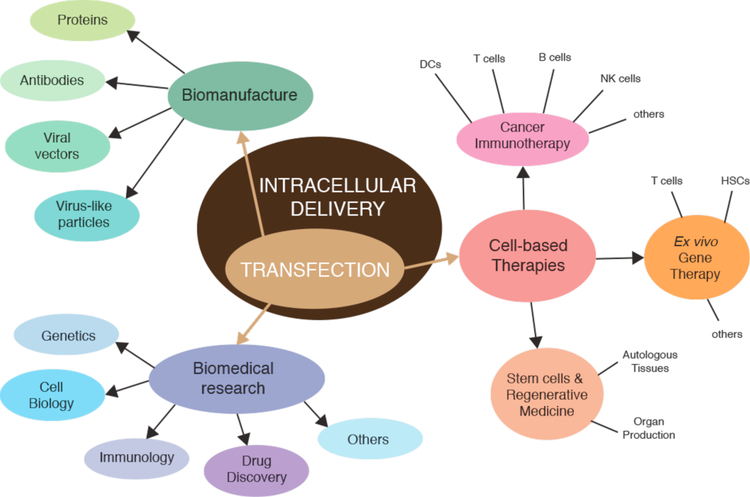
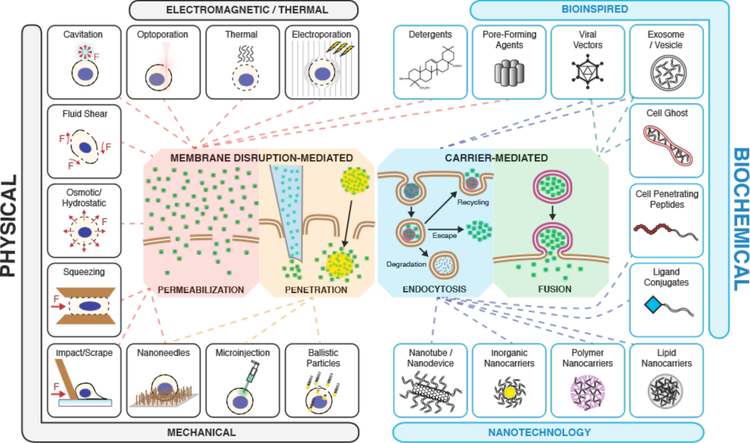
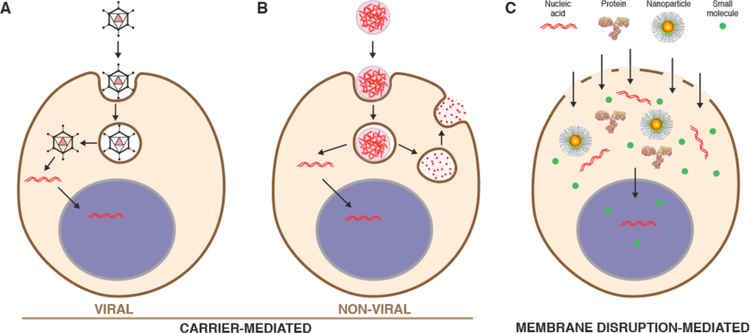
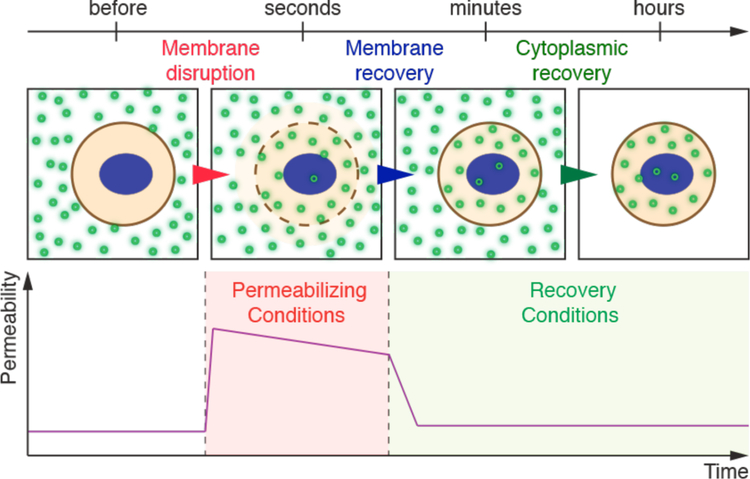
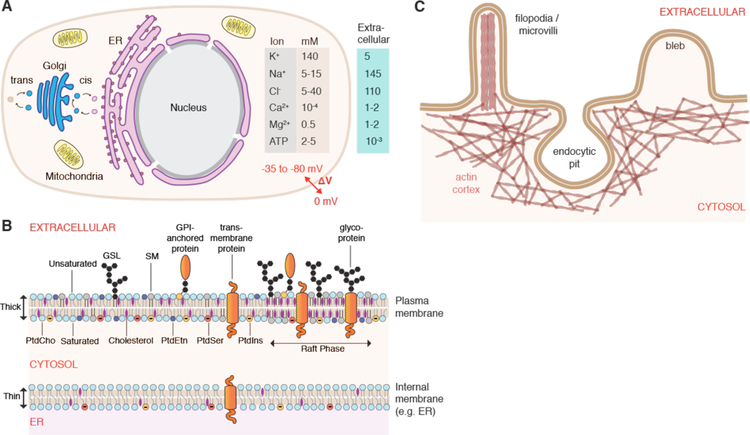
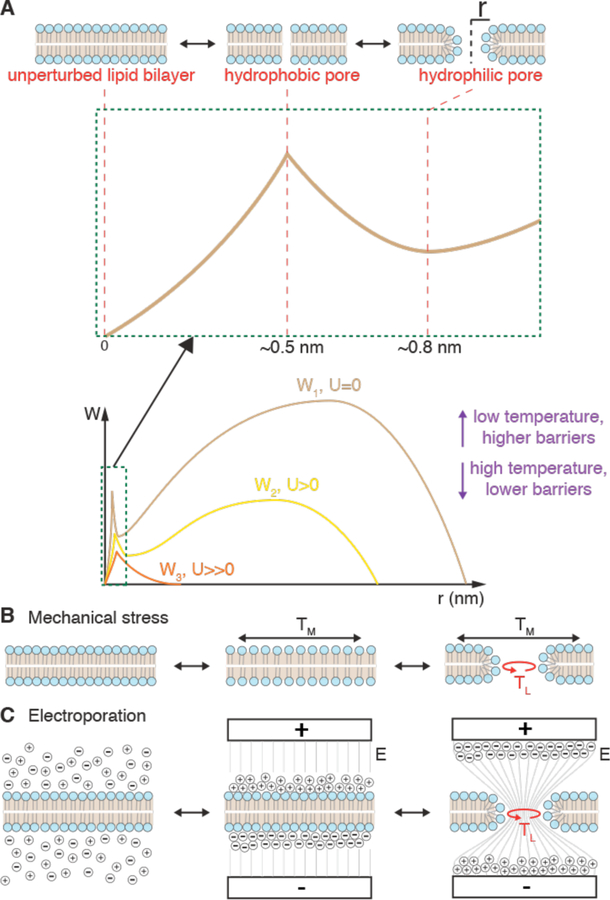
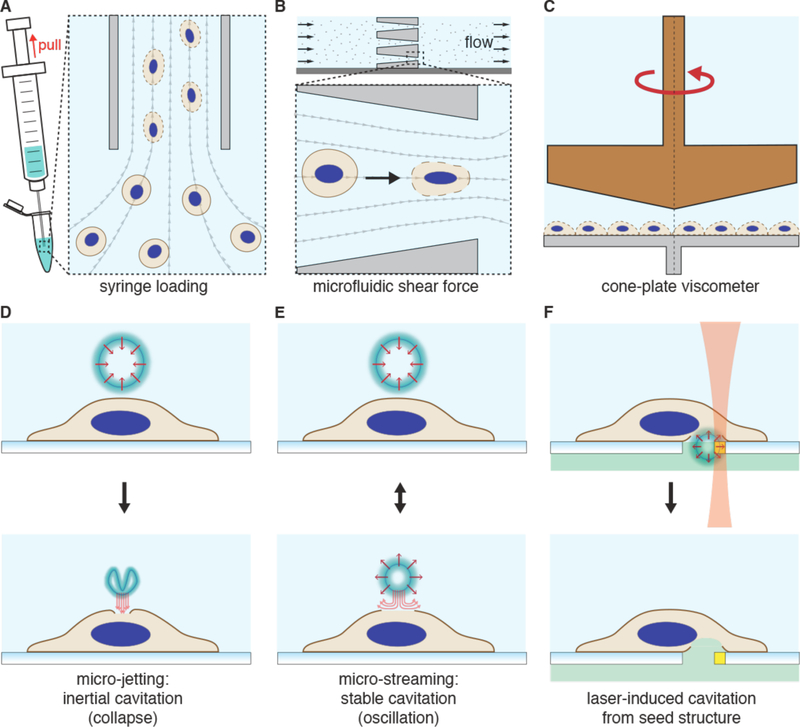
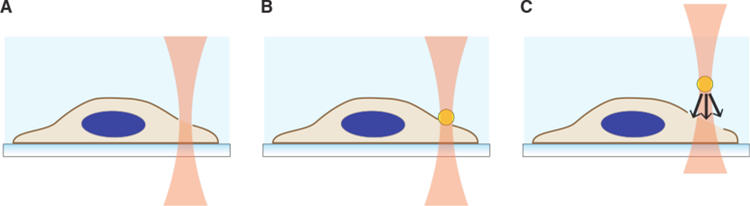
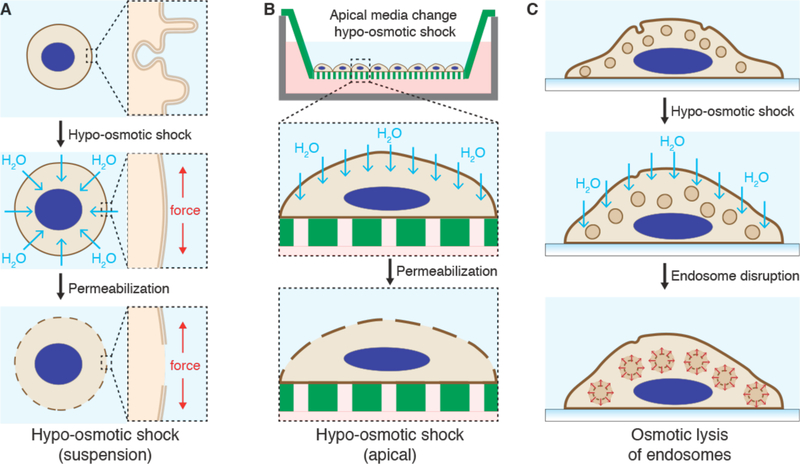
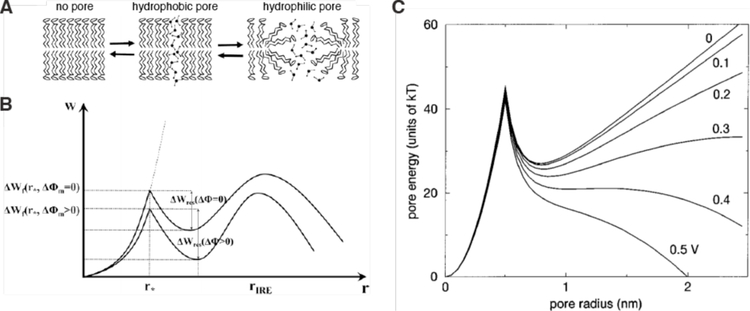
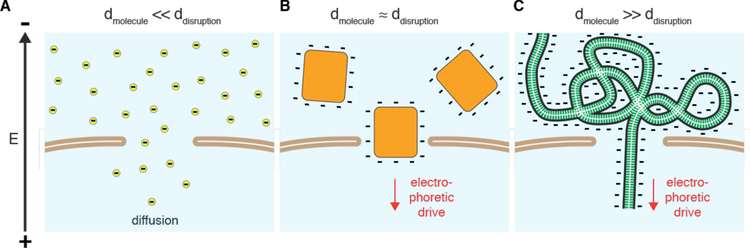
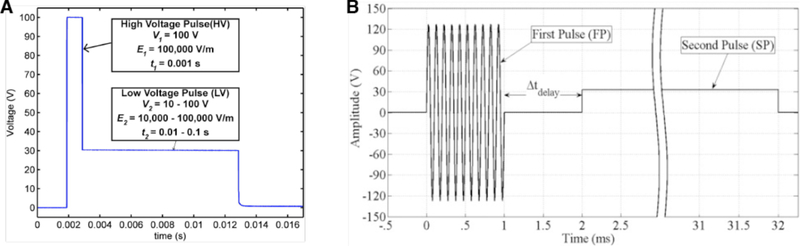
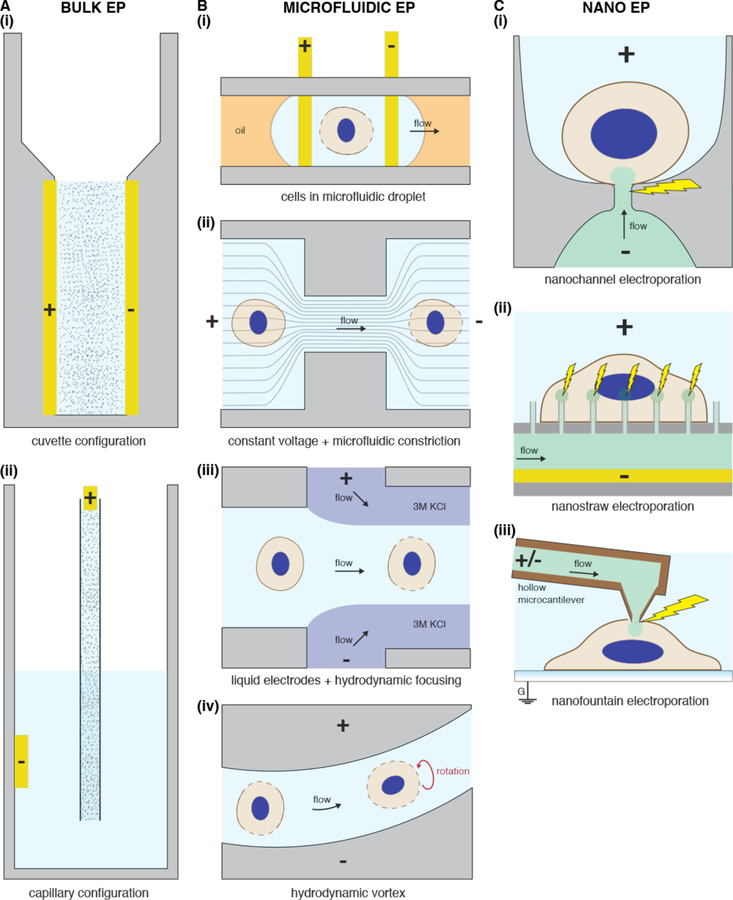
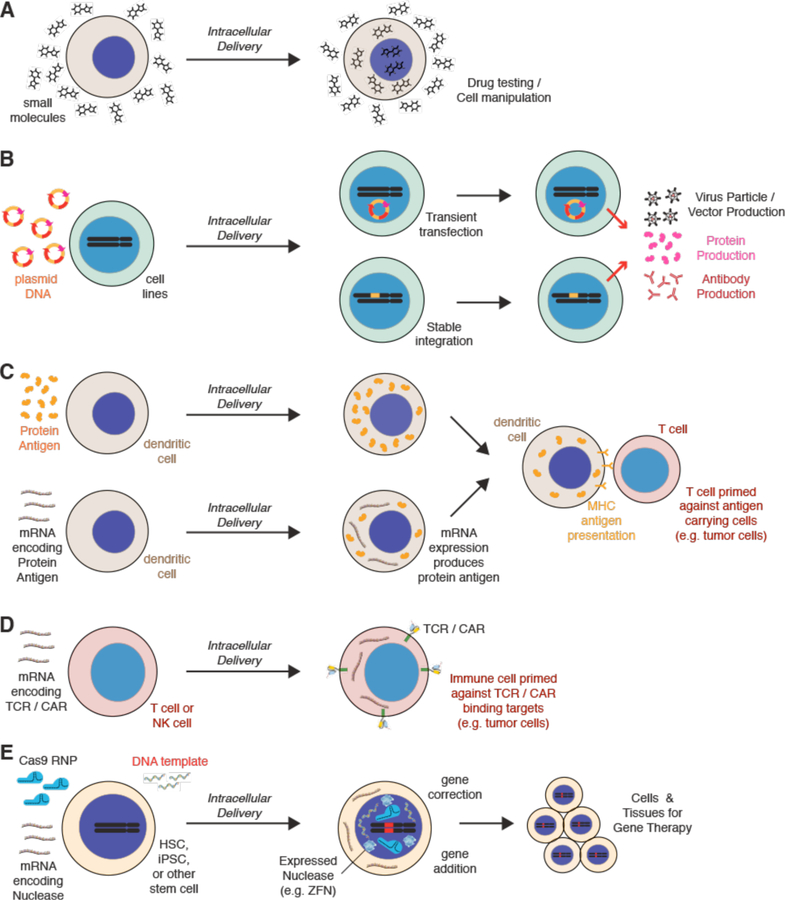
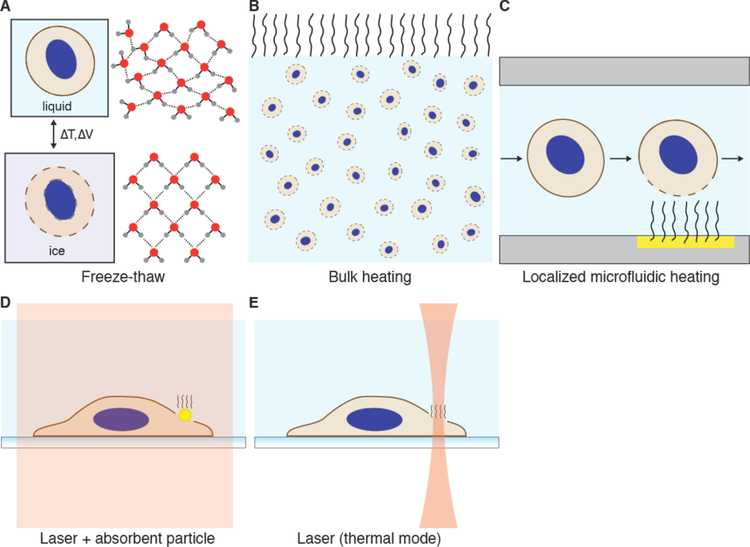
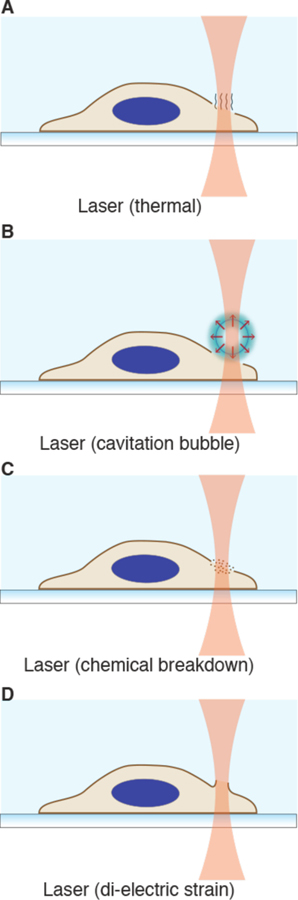
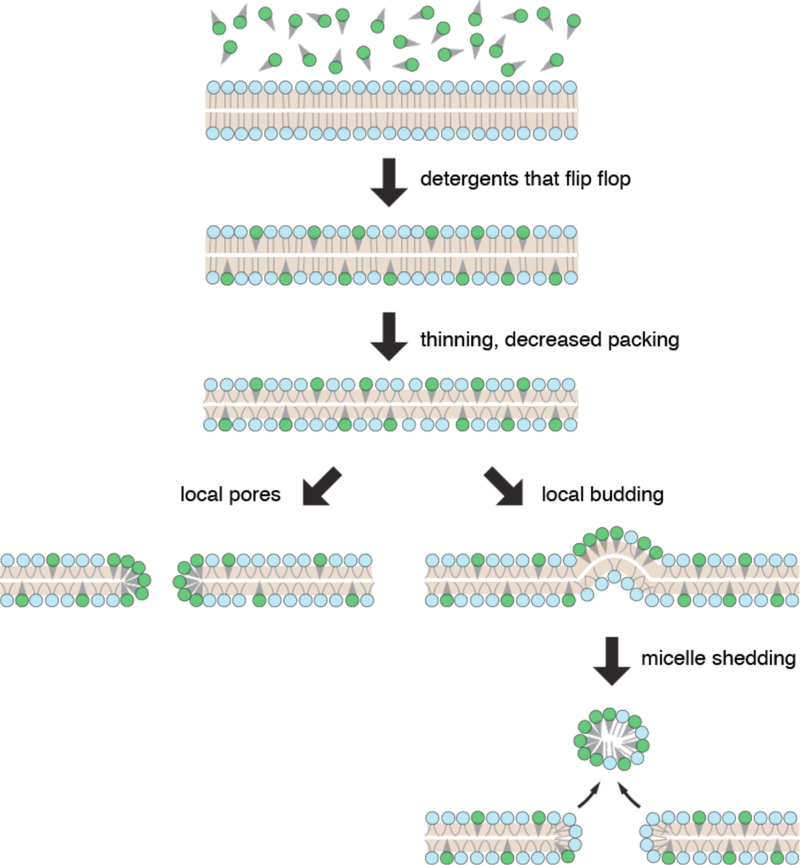
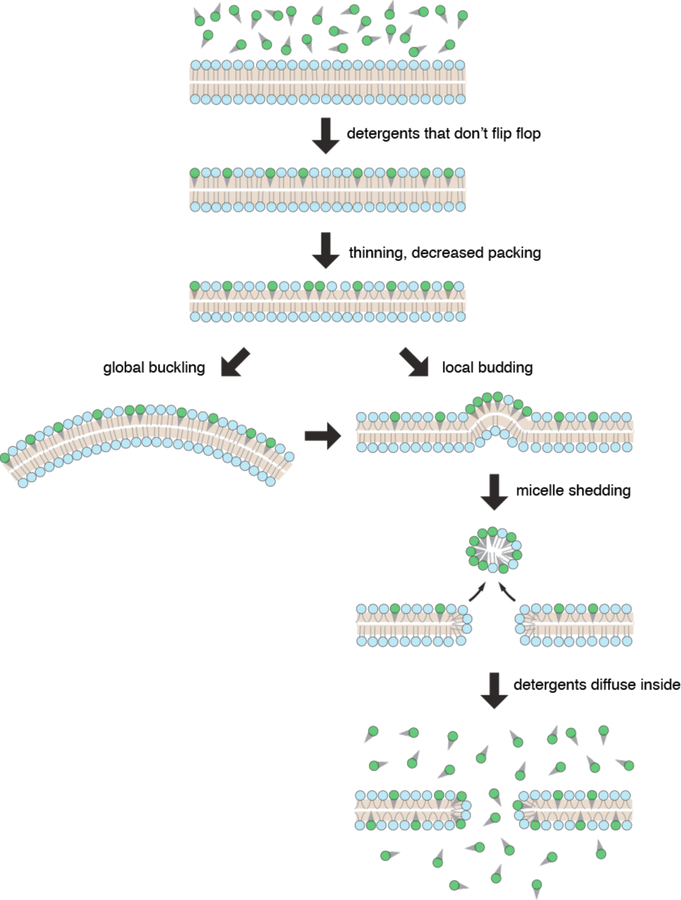
Intracellular delivery is a key step in biological research and has enabled decades of biomedical discoveries. It is also becoming increasingly important in industrial and medical applications ranging from biomanufacture to cell-based therapies. Here, we review techniques for membrane disruption-based intracellular delivery from 1911 until the present. These methods achieve rapid, direct, and universal delivery of almost any cargo molecule or material that can be dispersed in solution. We start by covering the motivations for intracellular delivery and the challenges associated with the different cargo types-small molecules, proteins/peptides, nucleic acids, synthetic nanomaterials, and large cargo. The review then presents a broad comparison of delivery strategies followed by an analysis of membrane disruption mechanisms and the biology of the cell response. We cover mechanical, electrical, thermal, optical, and chemical strategies of membrane disruption with a particular emphasis on their applications and challenges to implementation. Throughout, we highlight specific mechanisms of membrane disruption and suggest areas in need of further experimentation. We hope the concepts discussed in our review inspire scientists and engineers with further ideas to improve intracellular delivery.
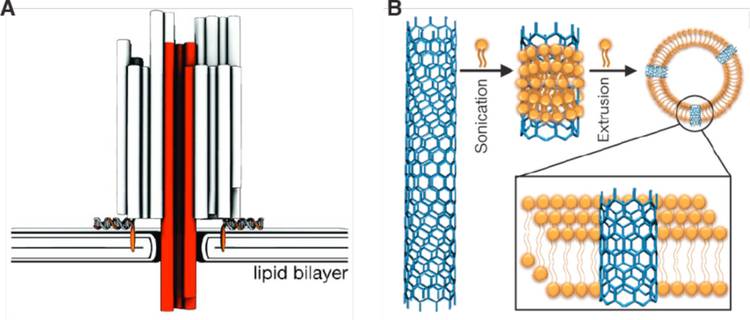
Figures
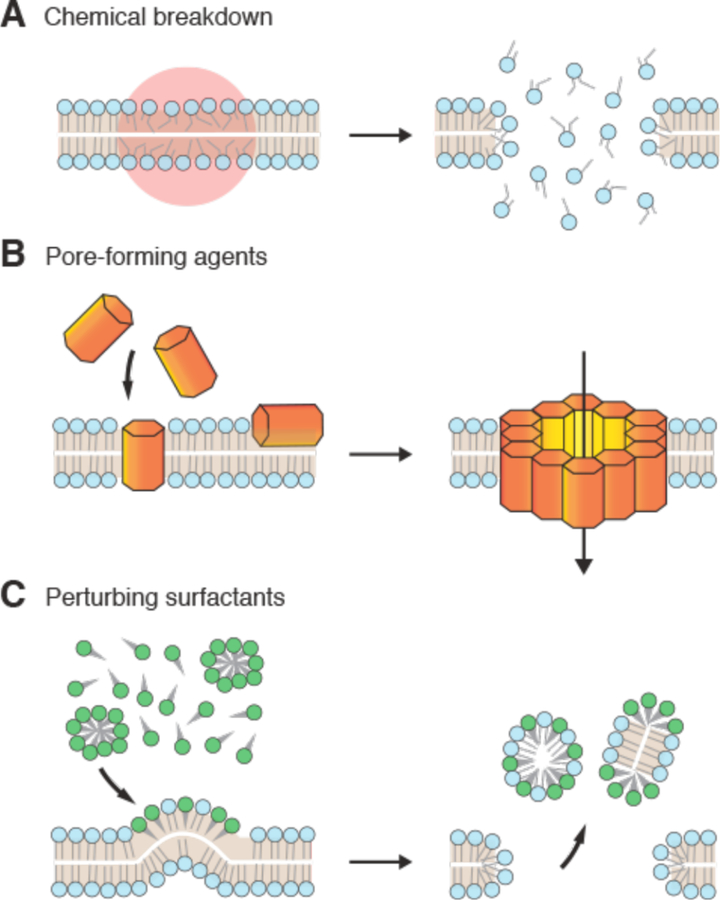
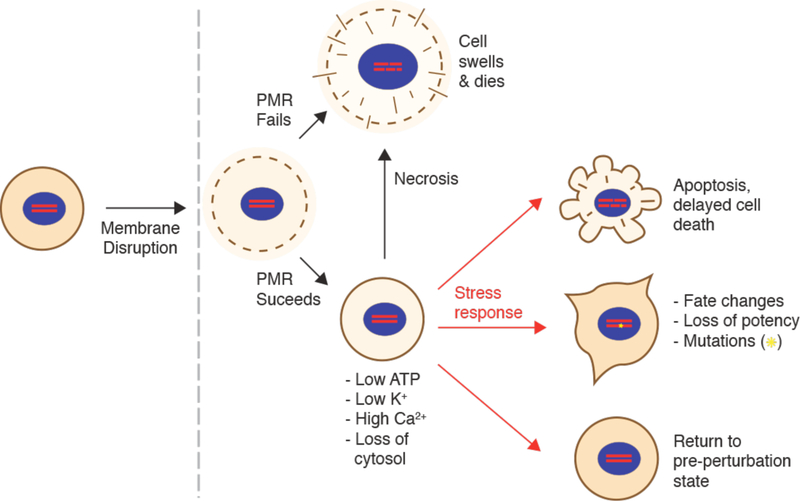
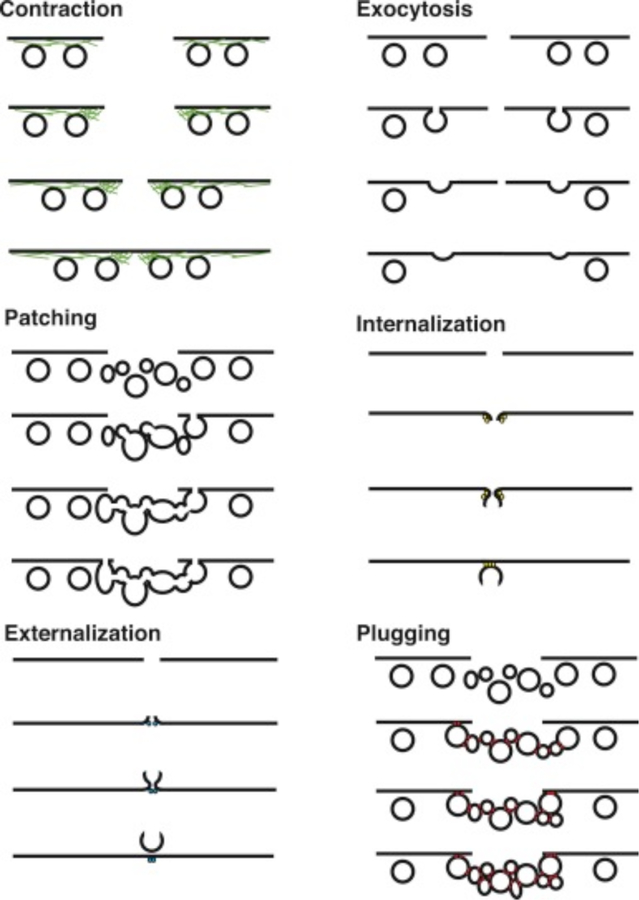
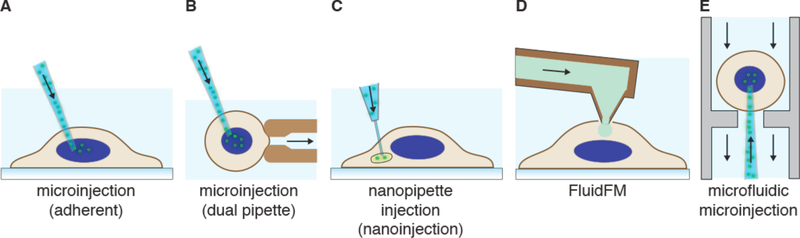
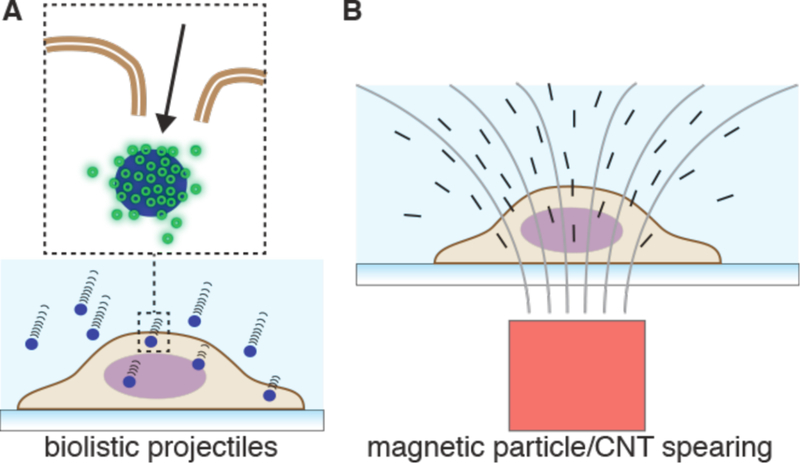
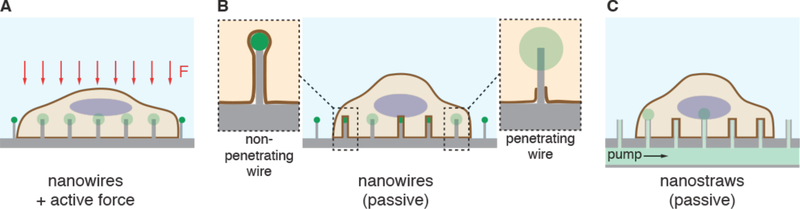
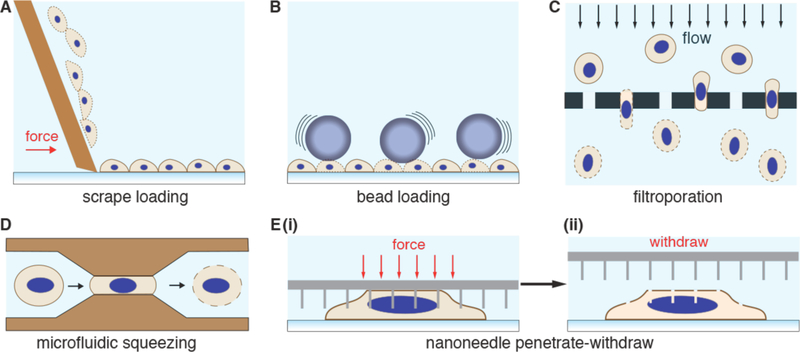
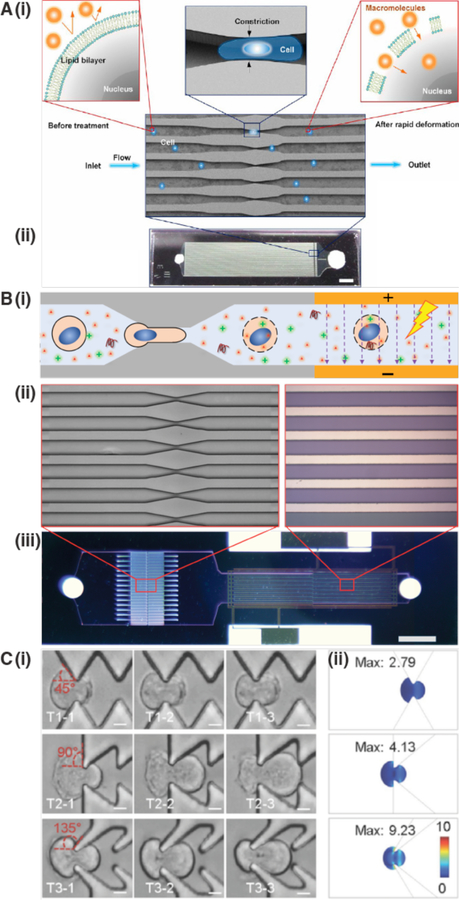
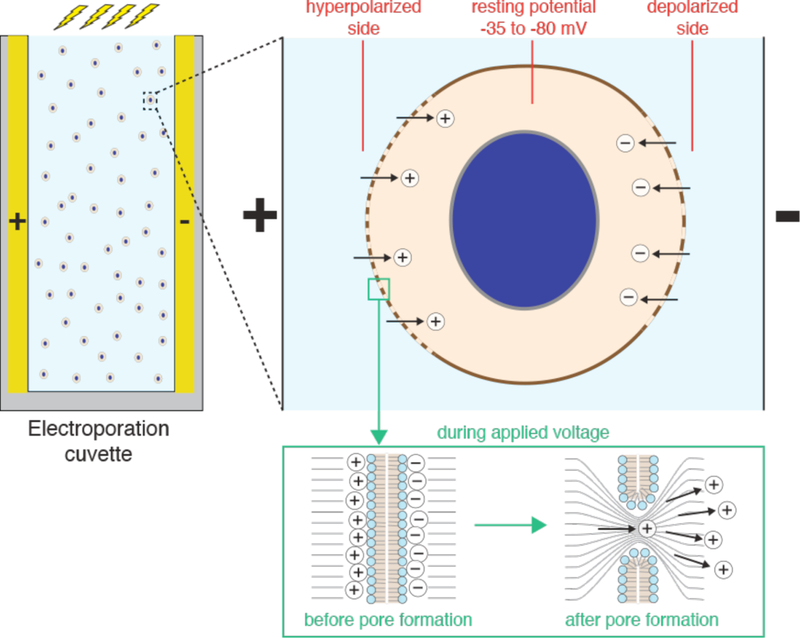
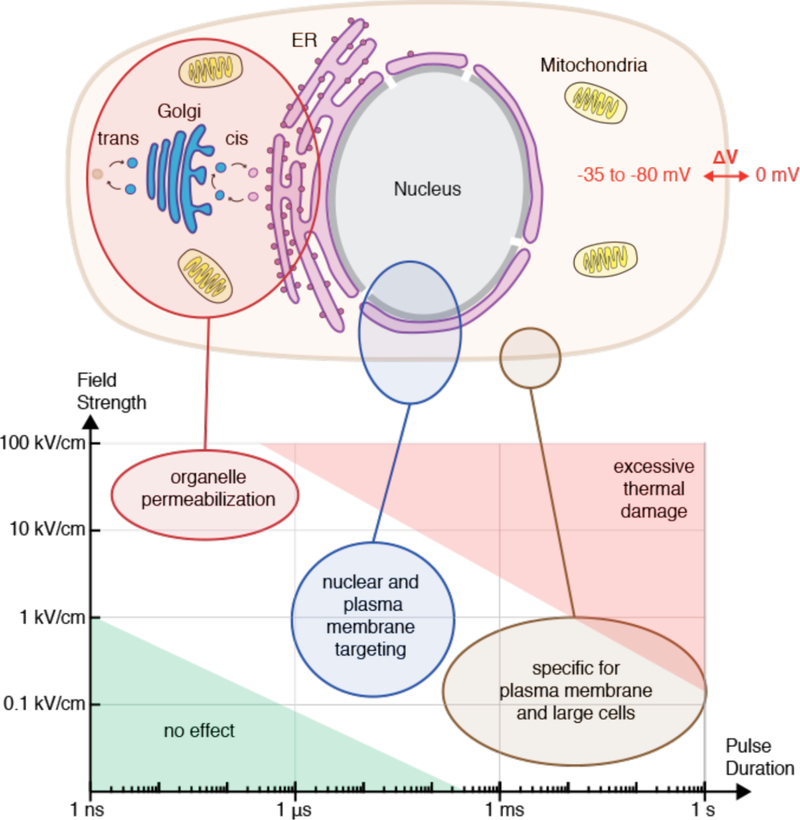
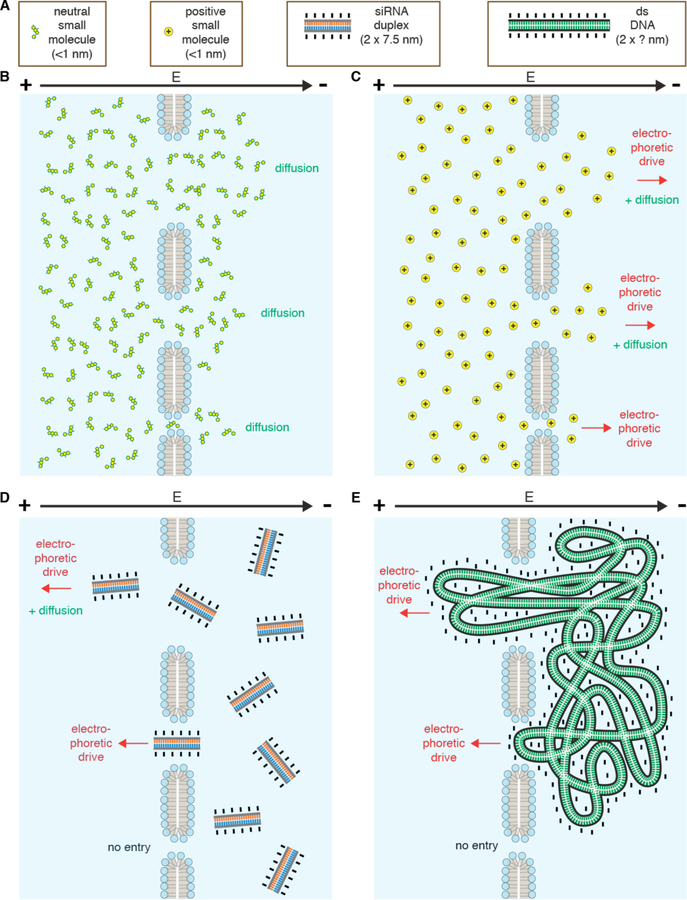
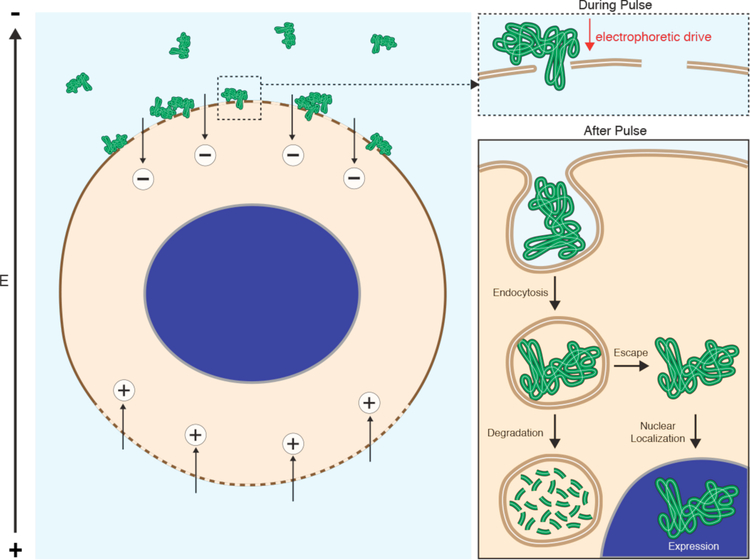

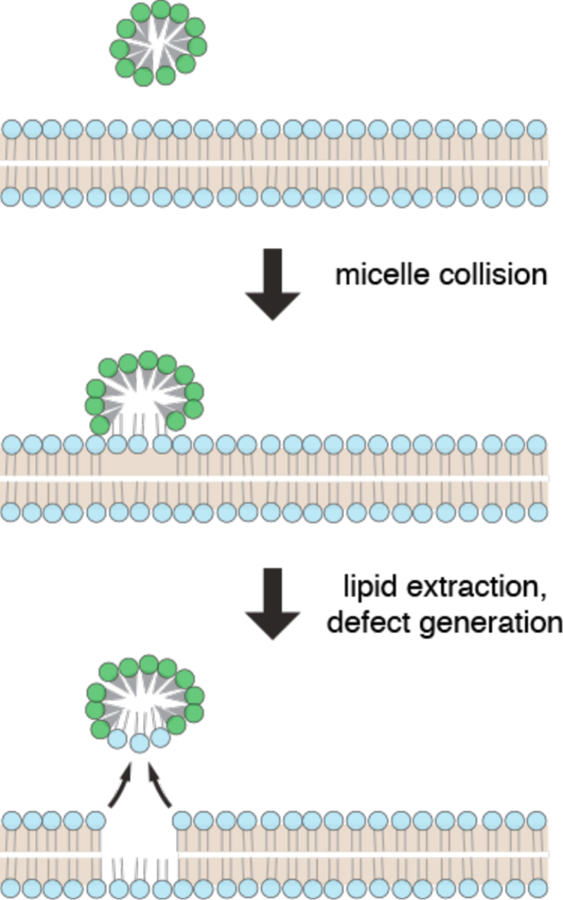
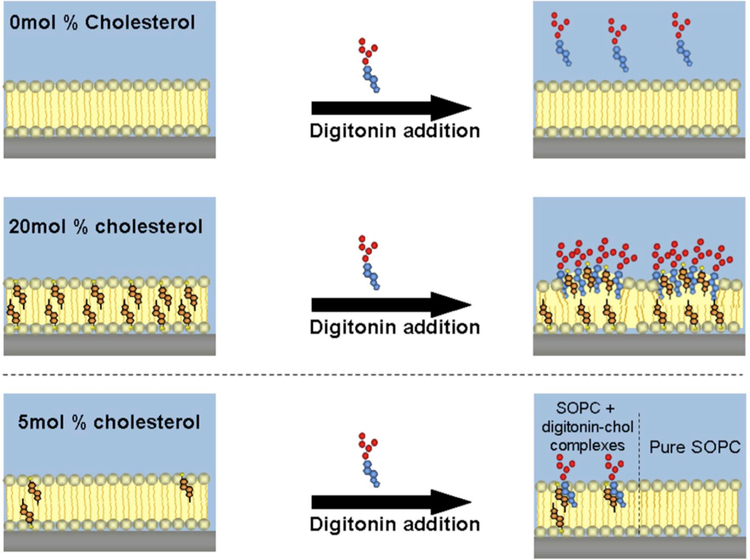
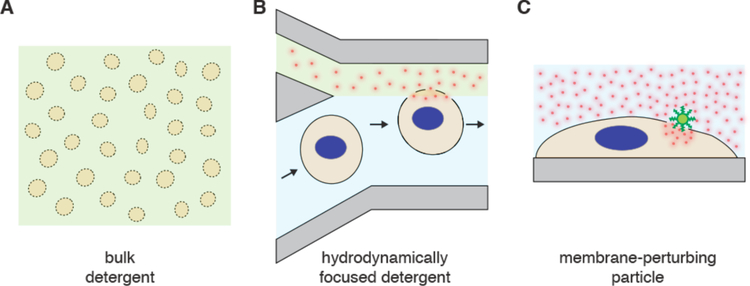
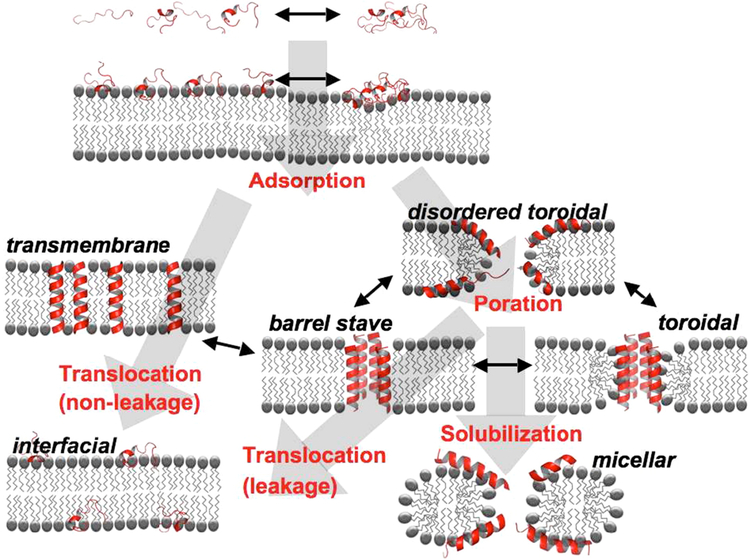
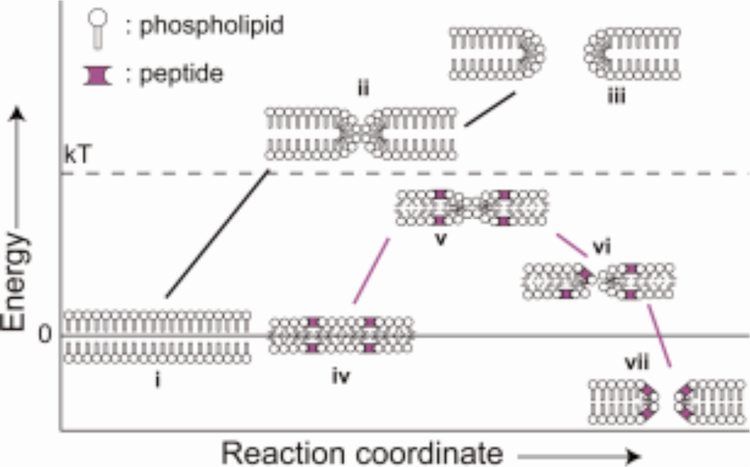
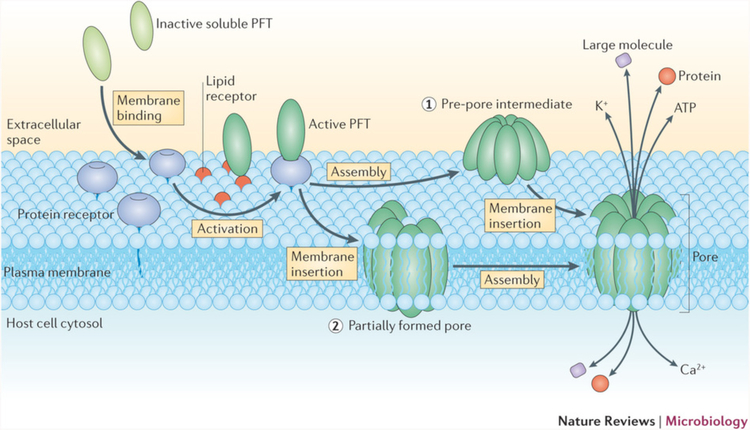
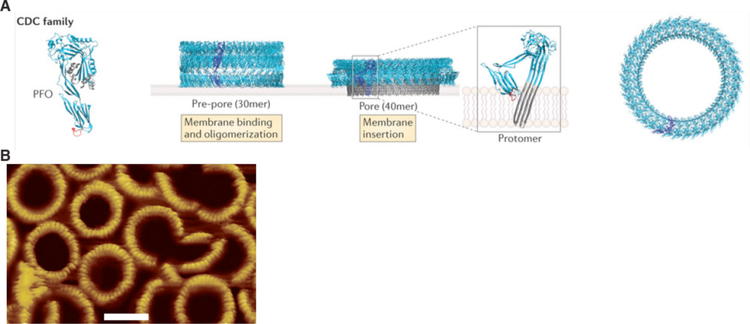
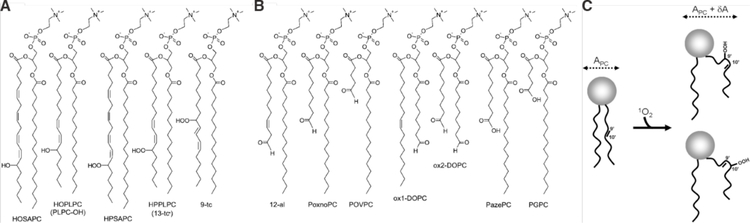
References
-
- Peer D; Karp JM; Hong S; Farokhzad OC; Margalit R; Langer R Nanocarriers as an emerging platform for cancer therapy. Nature nanotechnology 2007, 2 (12), 751. - PubMed
-
- Ganta S; Devalapally H; Shahiwala A; Amiji M A review of stimuli-responsive nanocarriers for drug and gene delivery. Journal of Controlled Release 2008, 126 (3), 187. - PubMed
-
- Mura S; Nicolas J; Couvreur P Stimuli-responsive nanocarriers for drug delivery. Nat Mater 2013, 12 (11), 991. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
