

Leveraging multiple transcriptome assembly methods for improved gene structure annotation
- PMID: 30052957
- PMCID: PMC6105091
- DOI: 10.1093/gigascience/giy093
Leveraging multiple transcriptome assembly methods for improved gene structure annotation
Abstract
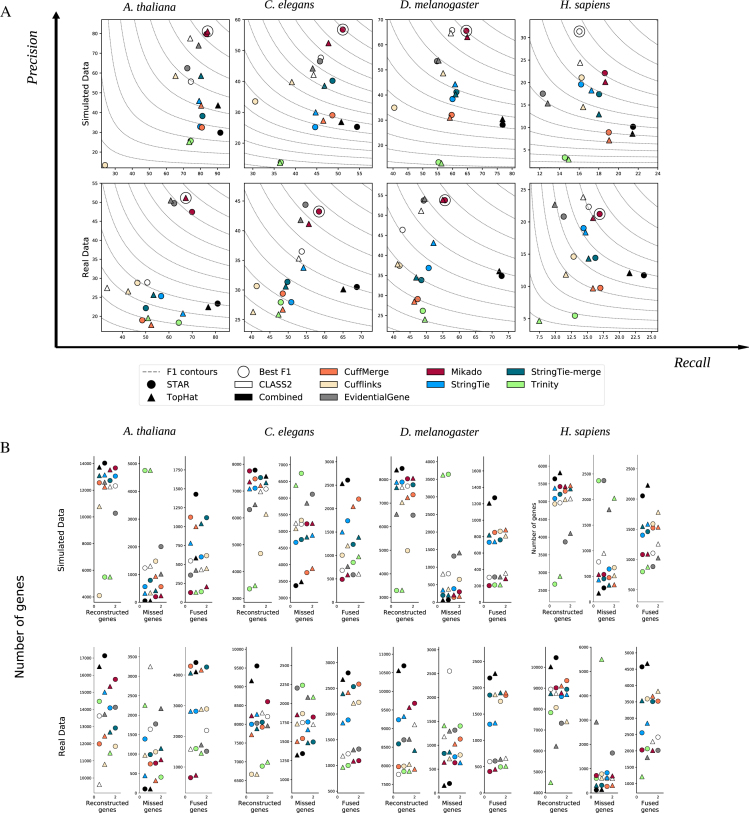
Background: The performance of RNA sequencing (RNA-seq) aligners and assemblers varies greatly across different organisms and experiments, and often the optimal approach is not known beforehand.
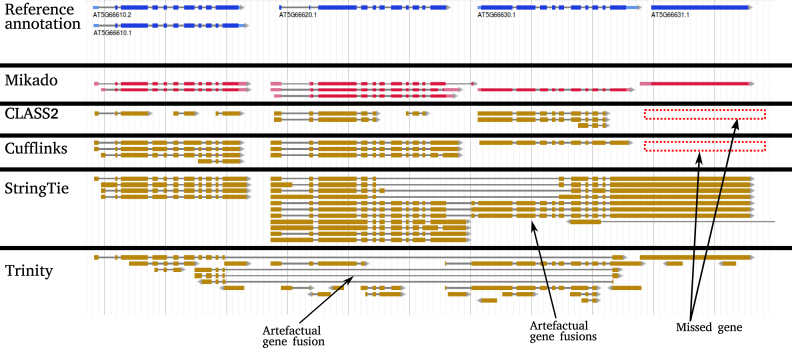
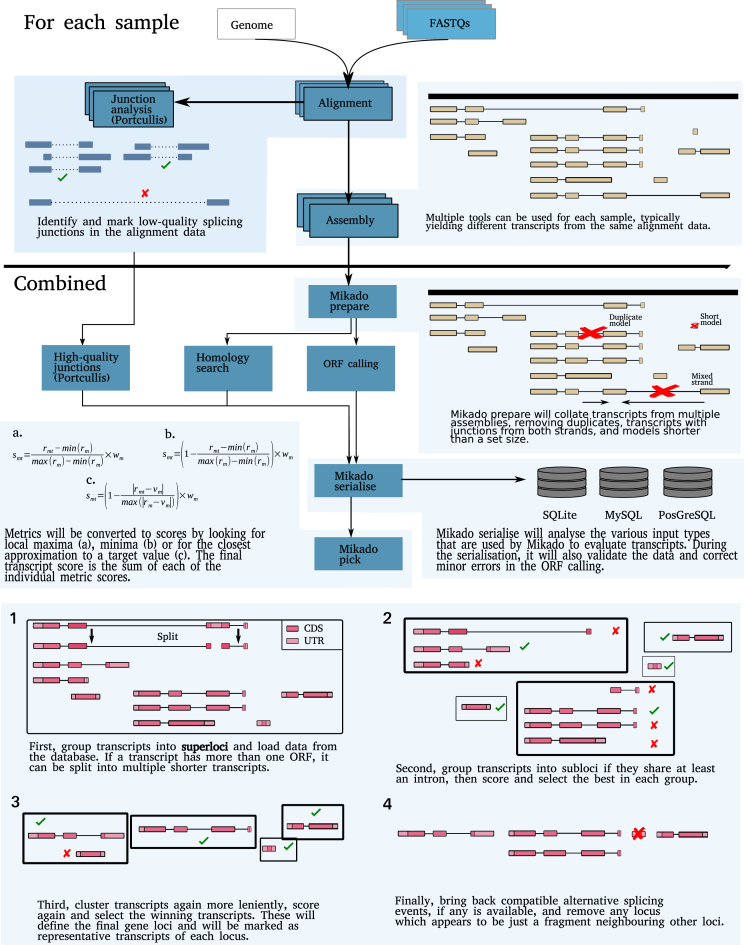
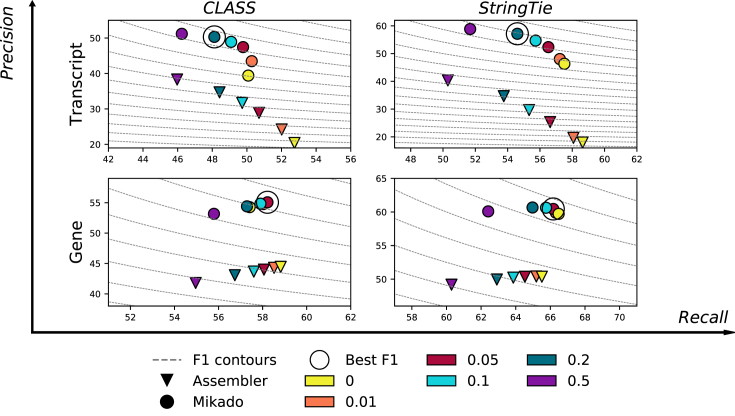
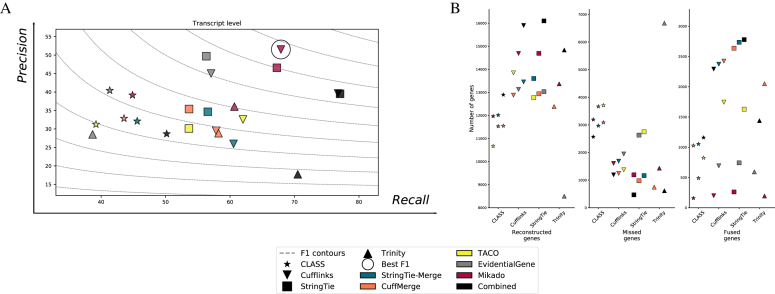
Results: Here, we show that the accuracy of transcript reconstruction can be boosted by combining multiple methods, and we present a novel algorithm to integrate multiple RNA-seq assemblies into a coherent transcript annotation. Our algorithm can remove redundancies and select the best transcript models according to user-specified metrics, while solving common artifacts such as erroneous transcript chimerisms.
Conclusions: We have implemented this method in an open-source Python3 and Cython program, Mikado, available on GitHub.
Figures
References
-
- Venturini L, Caim S, Kaithakottil GG et al. Mikado repository on GitHub; 2015. https://github.com/lucventurini/mikado/, Accessed 6 August, 2018.
-
- Bray NL, Pimentel H, Melsted P et al. Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology. 2016;34(5)525–527. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
- BBS/E/T/000PR9817/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/J003743/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/CSP1720/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/CCG1720/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
