

Muscle and adipose tissue insulin resistance: malady without mechanism?
- PMID: 30054342
- PMCID: PMC6795081
- DOI: 10.1194/jlr.R087510
Muscle and adipose tissue insulin resistance: malady without mechanism?
Abstract
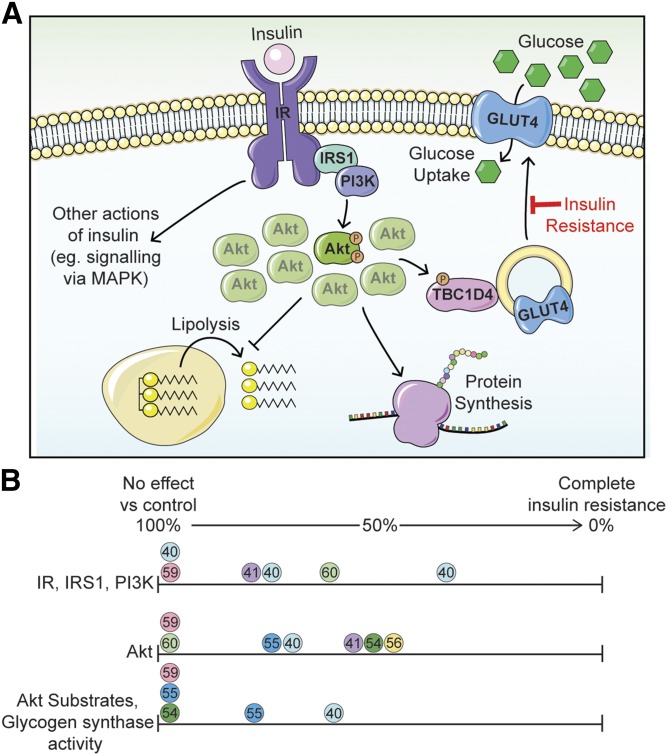
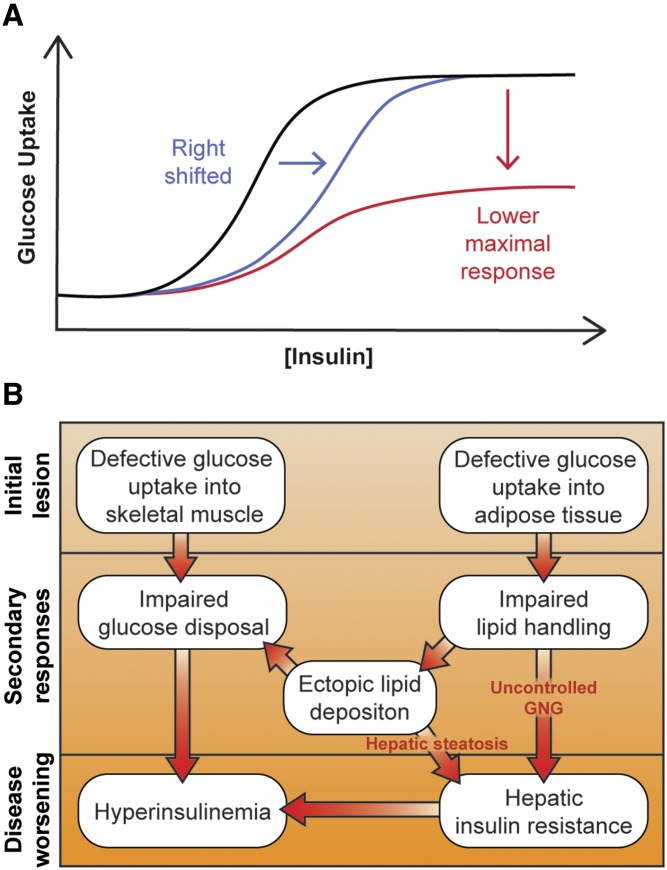
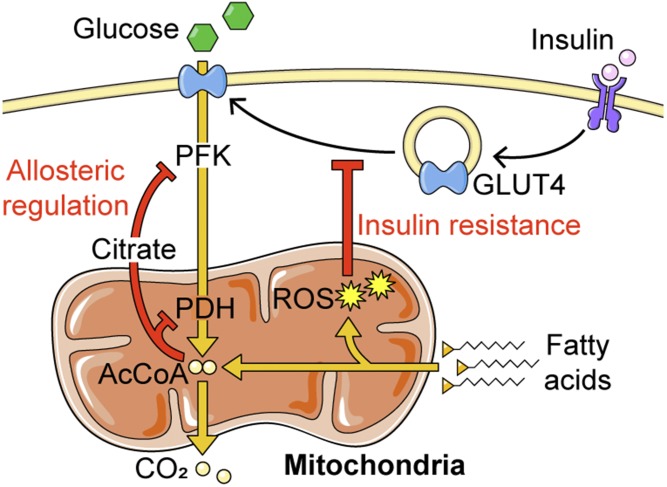
Insulin resistance is a major risk factor for numerous diseases, including type 2 diabetes and cardiovascular disease. These disorders have dramatically increased in incidence with modern life, suggesting that excess nutrients and obesity are major causes of "common" insulin resistance. Despite considerable effort, the mechanisms that contribute to common insulin resistance are not resolved. There is universal agreement that extracellular perturbations, such as nutrient excess, hyperinsulinemia, glucocorticoids, or inflammation, trigger intracellular stress in key metabolic target tissues, such as muscle and adipose tissue, and this impairs the ability of insulin to initiate its normal metabolic actions in these cells. Here, we present evidence that the impairment in insulin action is independent of proximal elements of the insulin signaling pathway and is likely specific to the glucoregulatory branch of insulin signaling. We propose that many intracellular stress pathways act in concert to increase mitochondrial reactive oxygen species to trigger insulin resistance. We speculate that this may be a physiological pathway to conserve glucose during specific states, such as fasting, and that, in the presence of chronic nutrient excess, this pathway ultimately leads to disease. This review highlights key points in this pathway that require further research effort.
Keywords: glucose transporter type 4; insulin signaling; mitochondria; oxidants.
Copyright © 2019 Fazakerley et al.
Figures
References
-
- Abdul-Ghani M. A., Tripathy D., and DeFronzo R. A.. 2006. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 29: 1130–1139. - PubMed
-
- Meyer C., Pimenta W., Woerle H. J., Van Haeften T., Szoke E., Mitrakou A., and Gerich J.. 2006. Different mechanisms for impaired fasting glucose and impaired postprandial glucose tolerance in humans. Diabetes Care. 29: 1909–1914. - PubMed
-
- Chen D. L., Liess C., Poljak A., Xu A., Zhang J., Thoma C., Trenell M., Milner B., Jenkins A. B., Chisholm D. J., et al. . 2015. Phenotypic characterization of insulin-resistant and insulin-sensitive obesity. J. Clin. Endocrinol. Metab. 100: 4082–4091. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
