

Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes
- PMID: 30054458
- PMCID: PMC6063971
- DOI: 10.1038/s41467-018-04951-w
Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes
Abstract
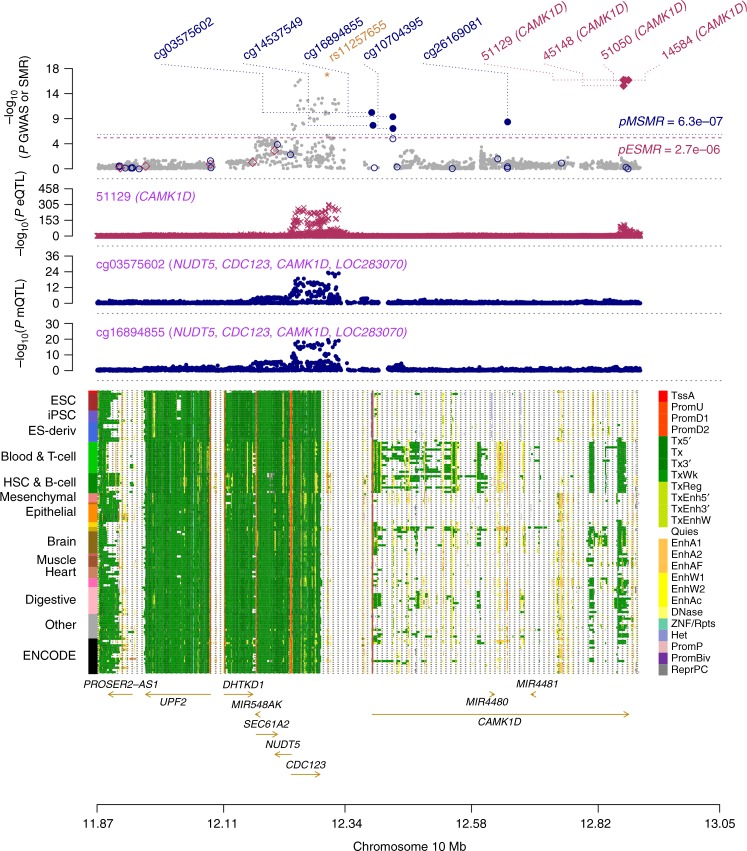
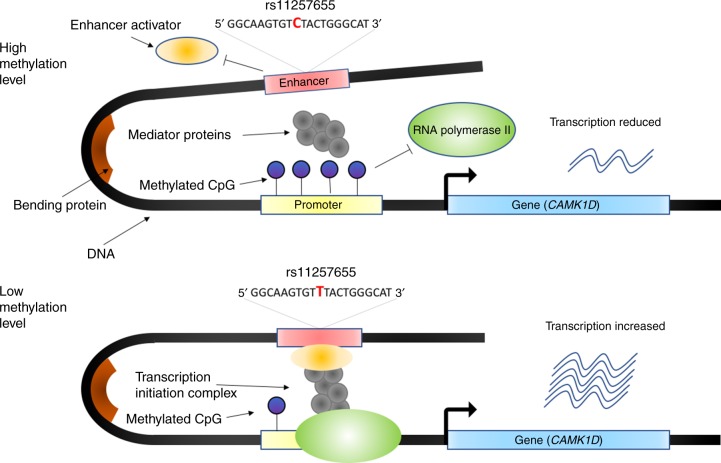
Type 2 diabetes (T2D) is a very common disease in humans. Here we conduct a meta-analysis of genome-wide association studies (GWAS) with ~16 million genetic variants in 62,892 T2D cases and 596,424 controls of European ancestry. We identify 139 common and 4 rare variants associated with T2D, 42 of which (39 common and 3 rare variants) are independent of the known variants. Integration of the gene expression data from blood (n = 14,115 and 2765) with the GWAS results identifies 33 putative functional genes for T2D, 3 of which were targeted by approved drugs. A further integration of DNA methylation (n = 1980) and epigenomic annotation data highlight 3 genes (CAMK1D, TP53INP1, and ATP5G1) with plausible regulatory mechanisms, whereby a genetic variant exerts an effect on T2D through epigenetic regulation of gene expression. Our study uncovers additional loci, proposes putative genetic regulatory mechanisms for T2D, and provides evidence of purifying selection for T2D-associated variants.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
