

The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis
- PMID: 30055613
- PMCID: PMC6064050
- DOI: 10.1186/s12931-018-0845-5
The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis
Abstract
Background: Since 2009, IPF patients across Europe are recruited into the eurIPFreg, providing epidemiological data and biomaterials for translational research.
Methods: The registry data are based on patient and physician baseline and follow-up questionnaires, comprising 1700 parameters. The mid- to long-term objectives of the registry are to provide clues for a better understanding of IPF phenotype sub-clusters, triggering factors and aggravating conditions, regional and environmental characteristics, and of disease behavior and management.
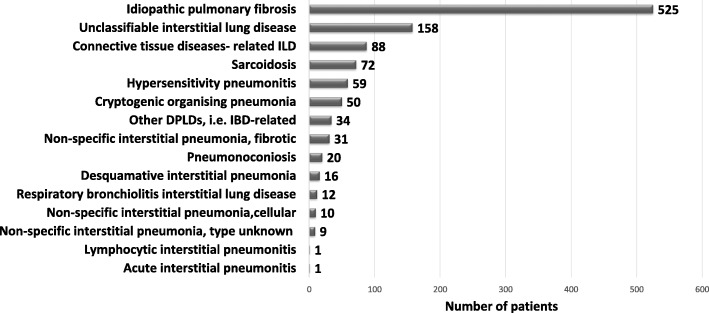
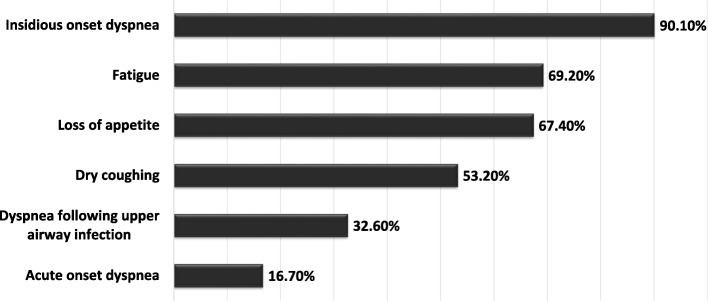
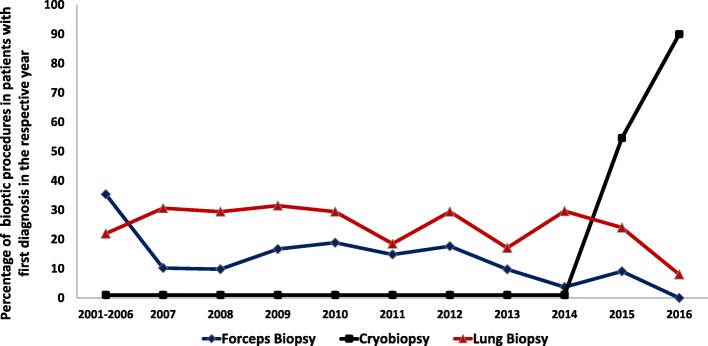
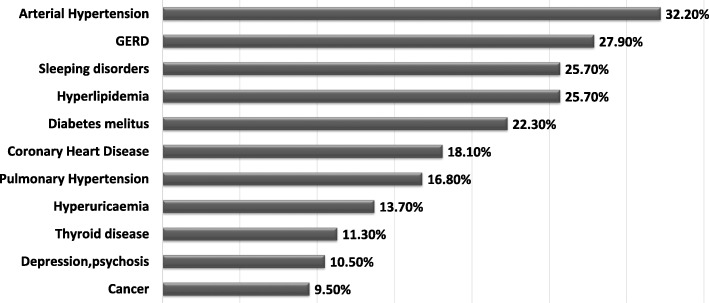
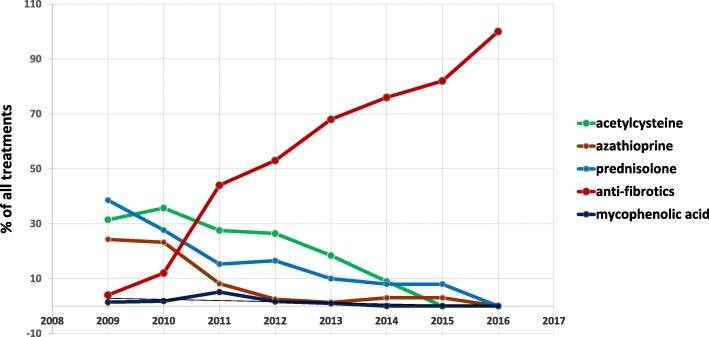
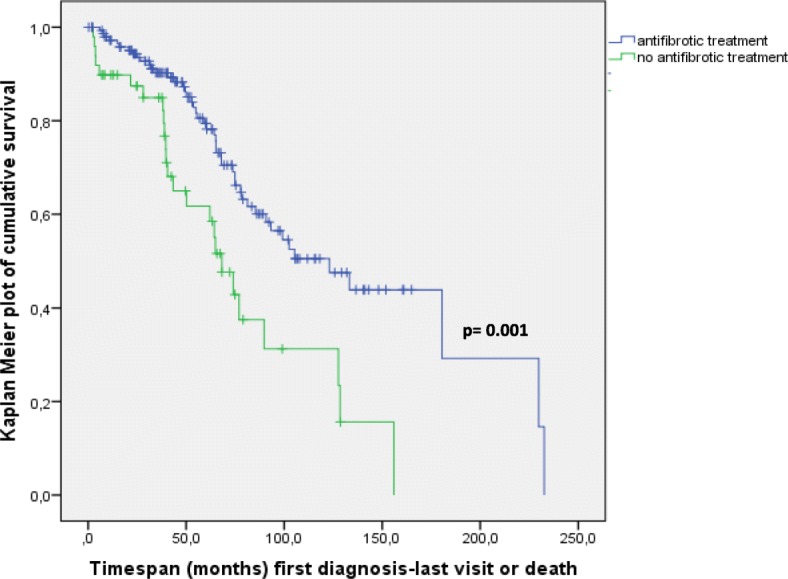
Results: This paper describes baseline data of 525 IPF subjects recruited from 11/2009 until 10/2016. IPF patients had a mean age of 68.1 years, and seeked medical advice due to insidious dyspnea (90.1%), fatigue (69.2%), and dry coughing (53.2%). A surgical lung biopsy was performed in 32% in 2009, but in only 8% of the cases in 2016, possibly due to increased numbers of cryobiopsy. At the time of inclusion in the eurIPFreg, FVC was 68.4% ± 22.6% of predicted value, DLco ranged at 42.1% ± 17.8% of predicted value (mean value ± SD). Signs of pulmonary hypertension were found in 16.8%. Steroids, immunosuppressants and N-Acetylcysteine declined since 2009, and were replaced by antifibrotics, under which patients showed improved survival (p = 0.001).
Conclusions: Our data provide important insights into baseline characteristics, diagnostic and management changes as well as outcome data in European IPF patients over time.
Trial registration: The eurIPFreg and eurIPFbank are listed in ClinicalTrials.gov( NCT02951416 ).
Keywords: European registry for idiopathic pulmonary fibrosis (eurIPFreg); Idiopathic pulmonary fibrosis (IPF); Interstitial lung diseases (ILD).
Conflict of interest statement
Ethics approval and consent to participate
Both, eurIPFreg and eurIPFbank have also been reviewed and approved from institutional review boards in Germany (e.g. Ethics Committee of Justus-Liebig-University of Giessen; 111/08), France, Italy, Austria, Spain, Czech Republic, Hungary and the UK. The research was conducted strictly according to the principles of the Declaration of Helsinki. Patients were included into the registry upon having signed the informed consent. The eurIPFreg and eurIPFbank are listed in
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Jo HE, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian idiopathic pulmonary fibrosis registry. Eur Respir J. 2017;49(2). 10.1183/13993003.01592-2016. - PubMed
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
