

Recent advances in developing therapeutics for cystic fibrosis
- PMID: 30060192
- PMCID: PMC6061831
- DOI: 10.1093/hmg/ddy188
Recent advances in developing therapeutics for cystic fibrosis
Abstract
Despite hope that a cure was imminent when the causative gene was cloned nearly 30 years ago, cystic fibrosis (CF [MIM: 219700]) remains a life-shortening disease affecting more than 70 000 individuals worldwide. However, within the last 6 years the Food and Drug Administration's approval of Ivacaftor, the first drug that corrects the defective cystic fibrosis transmembrane conductance regulator protein [CFTR (MIM: 602421)] in patients with the G551D mutation, marks a watershed in the development of novel therapeutics for this devastating disease. Here we review recent progress in diverse research areas, which all focus on curing CF at the genetic, biochemical or physiological level. In the near future it seems probable that development of mutation-specific therapies will be the focus, since it is unlikely that any one approach will be efficient in correcting the more than 2000 disease-associated variants. We discuss the new drugs and combinations of drugs that either enhance delivery of misfolded CFTR protein to the cell membrane, where it functions as an ion channel, or that activate channel opening. Next we consider approaches to correct the causative genetic lesion at the DNA or RNA level, through repressing stop mutations and nonsense-mediated decay, modulating splice mutations, fixing errors by gene editing or using novel routes to gene replacement. Finally, we explore how modifier genes, loci elsewhere in the genome that modify CF disease severity, may be used to restore a normal phenotype. Progress in all of these areas has been dramatic, generating enthusiasm that CF may soon become a broadly treatable disease.
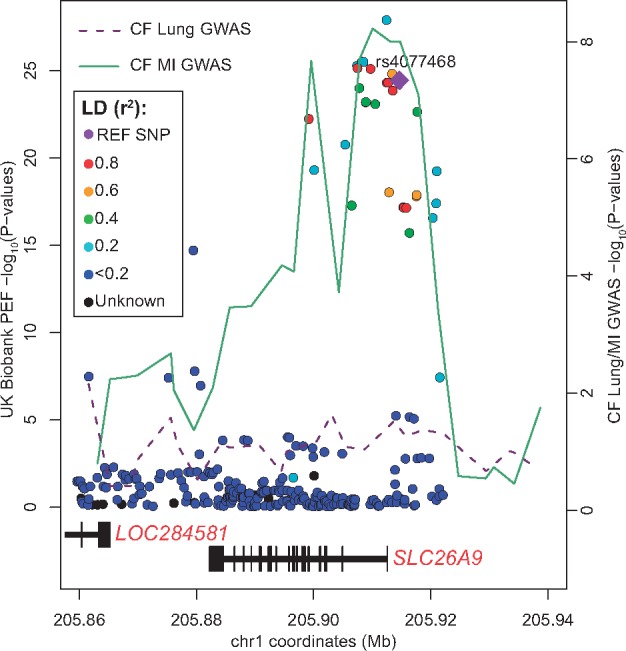
Figures
References
-
- Sawicki G.S., McKone E.F., Pasta D.J., Millar S.J., Wagener J.S., Johnson C.A., Konstan M.W. (2015) Sustained benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am. J. Respir. Crit. Care Med., 192, 836–842. - PubMed
-
- Taylor-Cousar J.L., Munck A., McKone E.F., van der Ent C.K., Moeller A., Simard C., Wang L.T., Ingenito E.P., McKee C., Lu Y.. et al. (2017) Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N. Engl. J. Med., 377, 2013–2023. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
