

The Tip of an Iceberg: Replication-Associated Functions of the Tumor Suppressor p53
- PMID: 30060597
- PMCID: PMC6115784
- DOI: 10.3390/cancers10080250
The Tip of an Iceberg: Replication-Associated Functions of the Tumor Suppressor p53
Abstract
The tumor suppressor p53 is a transcriptional factor broadly mutated in cancer. Most inactivating and gain of function mutations disrupt the sequence-specific DNA binding domain, which activates target genes. This is perhaps the main reason why most research has focused on the relevance of such transcriptional activity for the prevention or elimination of cancer cells. Notwithstanding, transcriptional regulation may not be the only mechanism underlying its role in tumor suppression and therapeutic responses. In the past, a direct role of p53 in DNA repair transactions that include the regulation of homologous recombination has been suggested. More recently, the localization of p53 at replication forks has been demonstrated and the effect of p53 on nascent DNA elongation has been explored. While some data sets indicate that the regulation of ongoing replication forks by p53 may be mediated by p53 targets such as MDM2 (murine double minute 2) and polymerase (POL) eta other evidences demonstrate that p53 is capable of controlling DNA replication by directly interacting with the replisome and altering its composition. In addition to discussing such findings, this review will also analyze the impact that p53-mediated control of ongoing DNA replication has on treatment responses and tumor suppressor abilities of this important anti-oncogene.
Keywords: MRE11; POL iota; POL teta; RAD52; ZRANB3; fork reversal; mutant p53; template switching; therapy resistance; translesion DNA synthesis.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
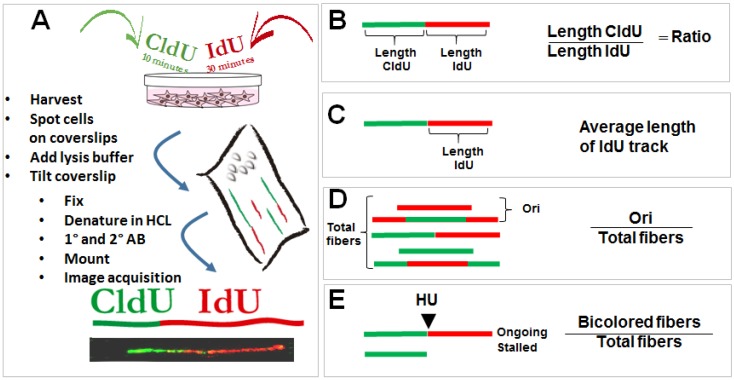
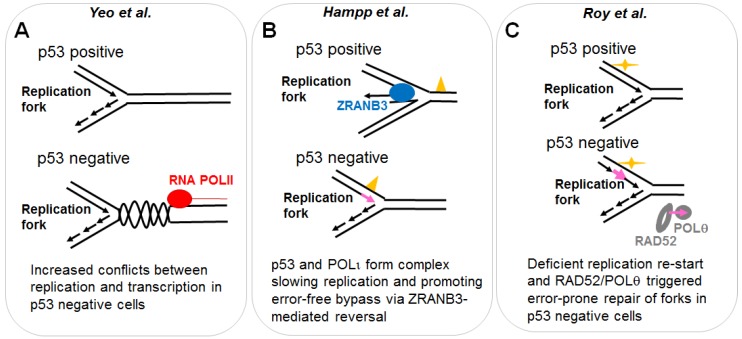
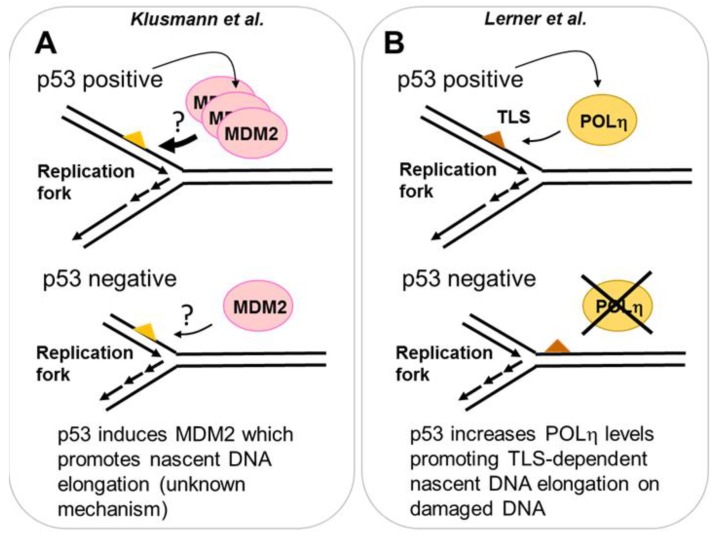
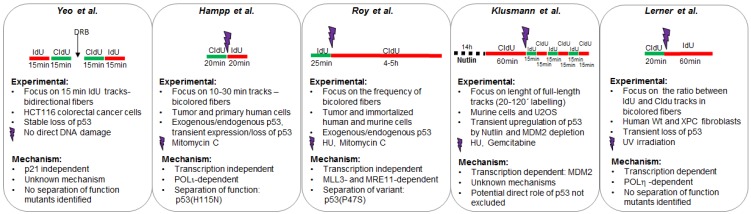
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
