

CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest
- PMID: 30061045
- PMCID: PMC6689321
- DOI: 10.1016/j.tcb.2018.07.002
CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest
Abstract
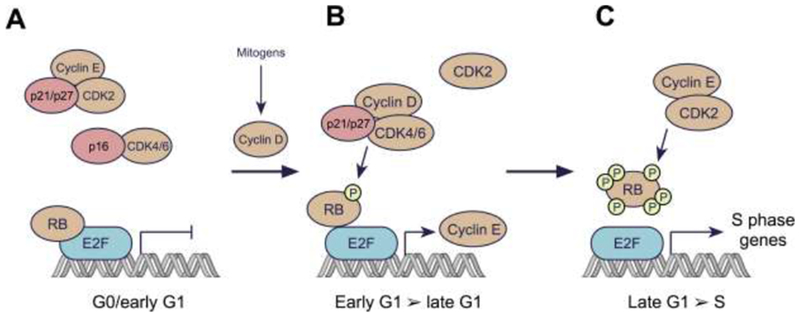
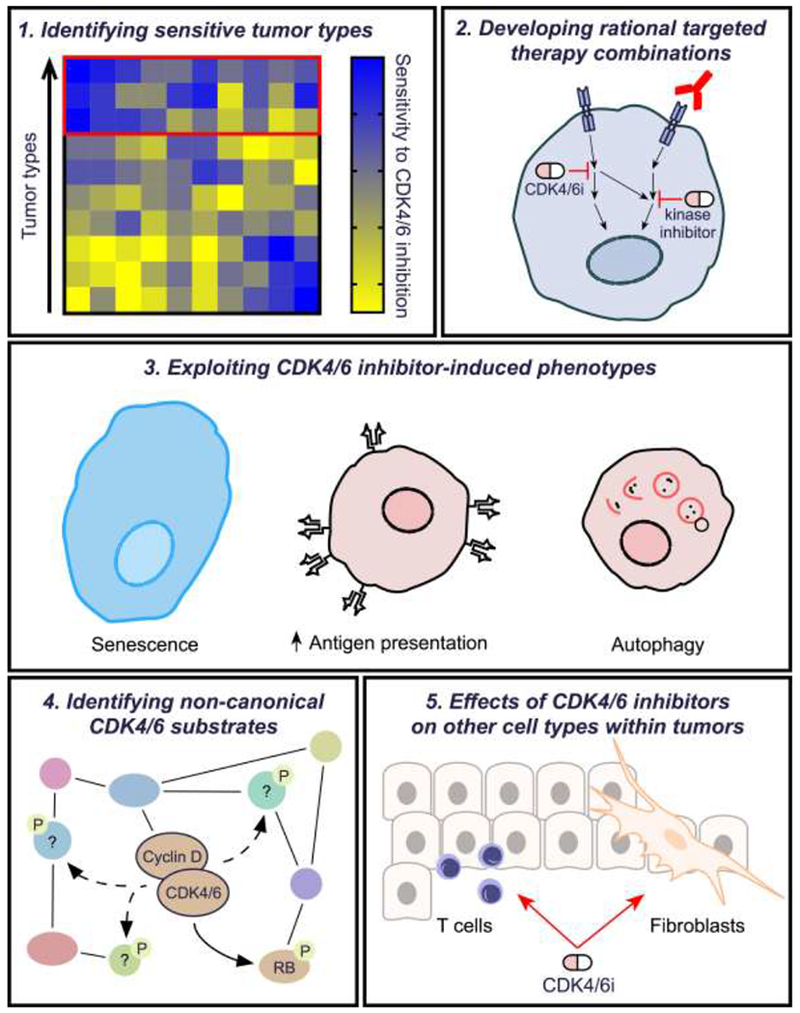
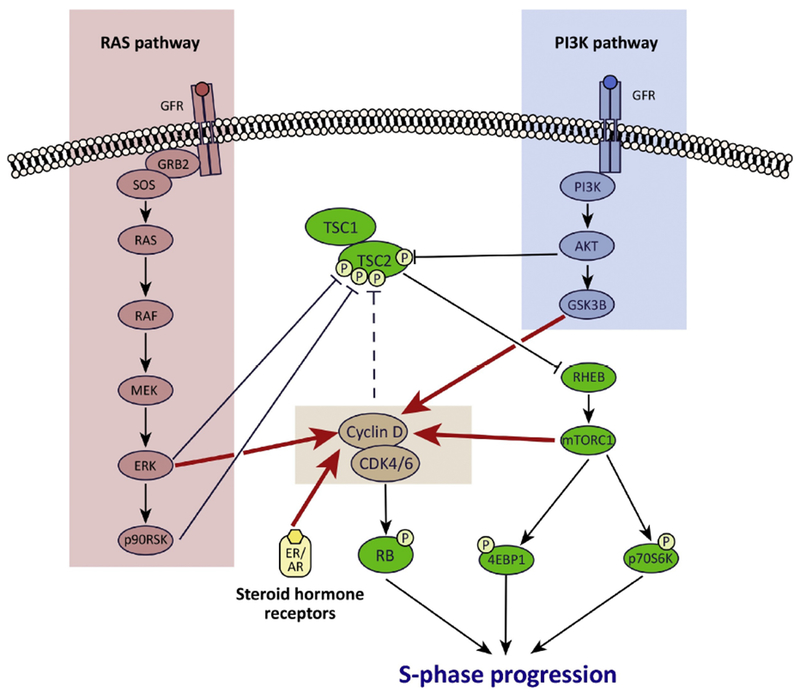
Pharmacologic inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6) have recently entered the therapeutic armamentarium of clinical oncologists, and show promising activity in patients with breast and other cancers. Although their chief mechanism of action is inhibition of retinoblastoma (RB) protein phosphorylation and thus the induction of cell cycle arrest, CDK4/6 inhibitors alter cancer cell biology in other ways that can also be leveraged for therapeutic benefit. These include modulation of mitogenic kinase signaling, induction of a senescence-like phenotype, and enhancement of cancer cell immunogenicity. We describe here the less-appreciated effects of CDK4/6 inhibitors on cancer cells, and suggest ways by which they might be exploited to enhance the benefits of these agents for cancer patients.
Keywords: CDK4/6; cell cycle; immunotherapy; targeted therapy.
Copyright © 2018. Published by Elsevier Ltd.
Figures
References
-
- Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5), 646–74. - PubMed
-
- Fry DW et al. (2004) Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 3 (11), 1427–38. - PubMed
-
- Kim S et al. (2014) LEE011: An orally bioavailable, selective small molecule inhibitor of CDK4/6-Reactivating Rb in cancer. Molecular Cancer Therapeutics 12 (PR02–PR02).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
