

Recurrent loss of heterozygosity correlates with clinical outcome in pancreatic neuroendocrine cancer
- PMID: 30062048
- PMCID: PMC6054670
- DOI: 10.1038/s41525-018-0058-3
Recurrent loss of heterozygosity correlates with clinical outcome in pancreatic neuroendocrine cancer
Abstract
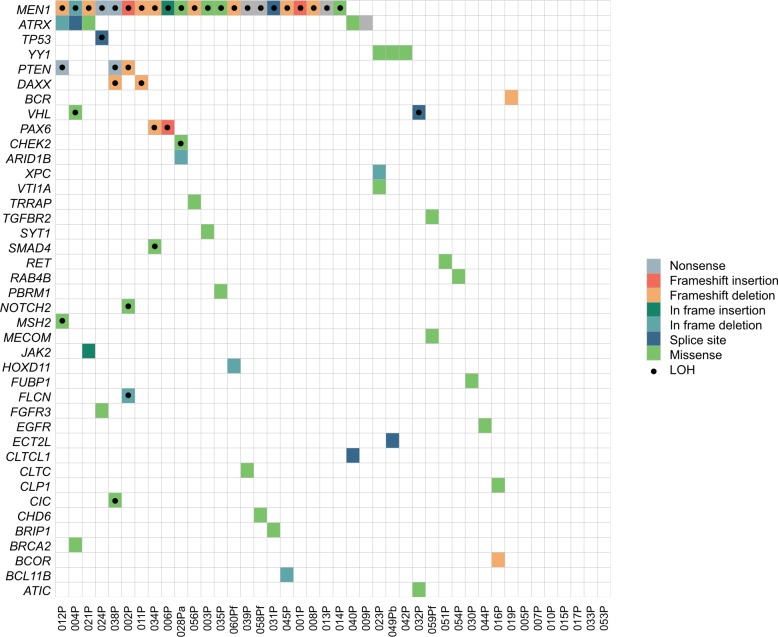
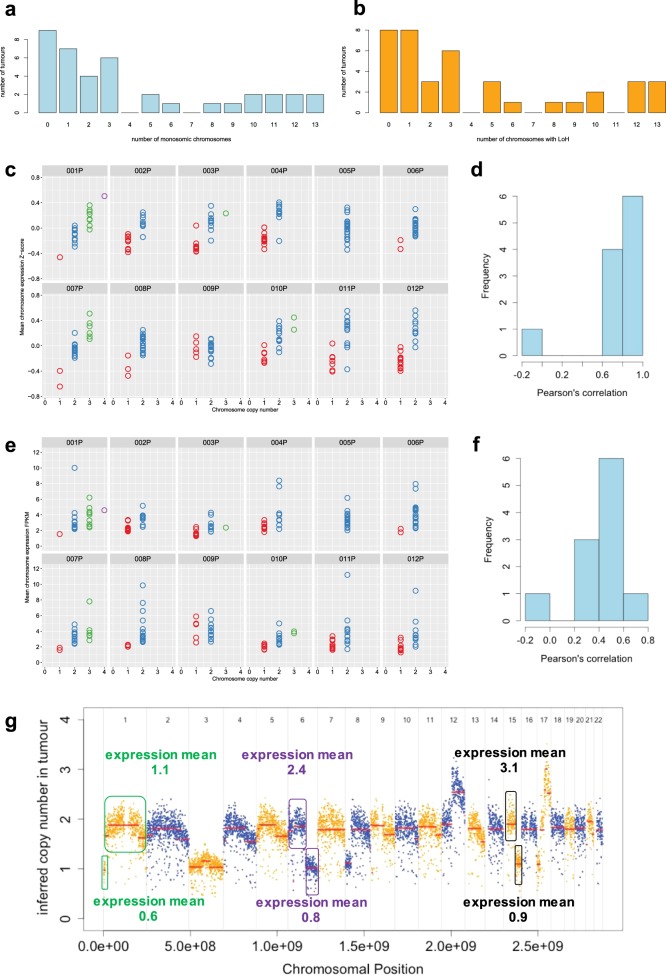
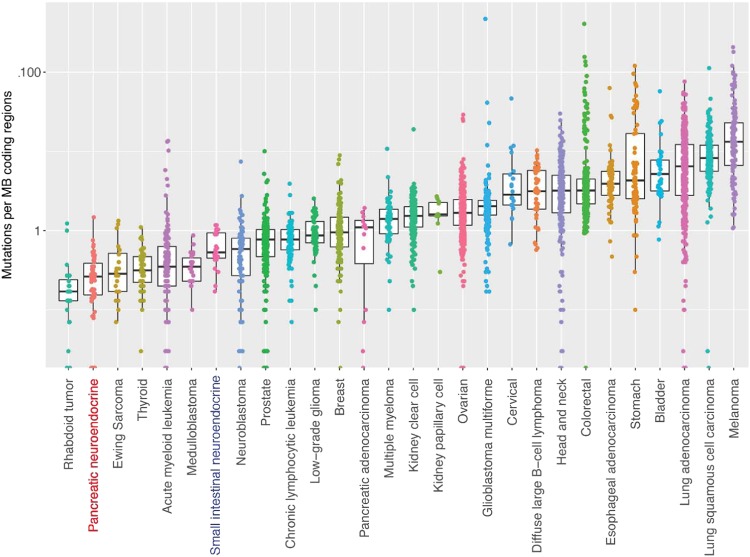
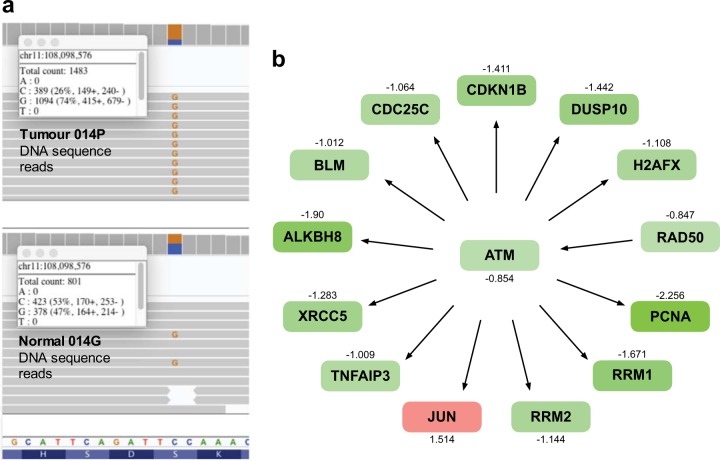
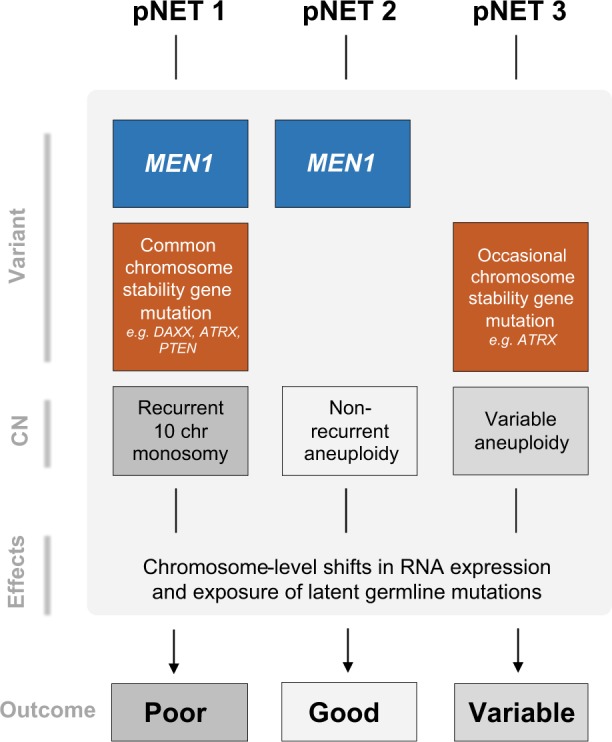
Pancreatic neuroendocrine tumors (pNETs) are uncommon cancers arising from pancreatic islet cells. Here we report the analysis of gene mutation, copy number, and RNA expression of 57 sporadic well-differentiated pNETs. pNET genomes are dominated by aneuploidy, leading to concordant changes in RNA expression at the level of whole chromosomes and chromosome segments. We observed two distinct patterns of somatic pNET aneuploidy that are associated with tumor pathology and patient prognosis. Approximately 26% of the patients in this series had pNETs with genomes characterized by recurrent loss of heterozygosity (LoH) of 10 specific chromosomes, accompanied by bi-allelic MEN1 inactivation and generally poor clinical outcome. Another ~40% of patients had pNETs that lacked this recurrent LoH pattern but had chromosome 11 LoH, bi-allelic MEN1 inactivation, and universally good clinical outcome. The somatic aneuploidy allowed pathogenic germline variants (e.g., ATM) to be expressed unopposed, with RNA expression patterns showing inactivation of downstream tumor suppressor pathways. No prognostic associations were found with tumor morphology, single gene mutation, or expression of RNAs reflecting the activity of immune, differentiation, proliferative or tumor suppressor pathways. In pNETs, single gene mutations appear to be less important than aneuploidy, with MEN1 the only statistically significant recurrently mutated driver gene. In addition, only one pNET in the series had clearly actionable single nucleotide variants (SNVs) (in PTEN and FLCN) confirmed by corroborating RNA expression changes. The two clinically relevant patterns of LoH described here define a novel oncogenic mechanism and a plausible route to genomic precision oncology for this tumor type.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
