

Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich's Ataxia Models
- PMID: 30065630
- PMCID: PMC6056642
- DOI: 10.3389/fncel.2018.00188
Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich's Ataxia Models
Abstract
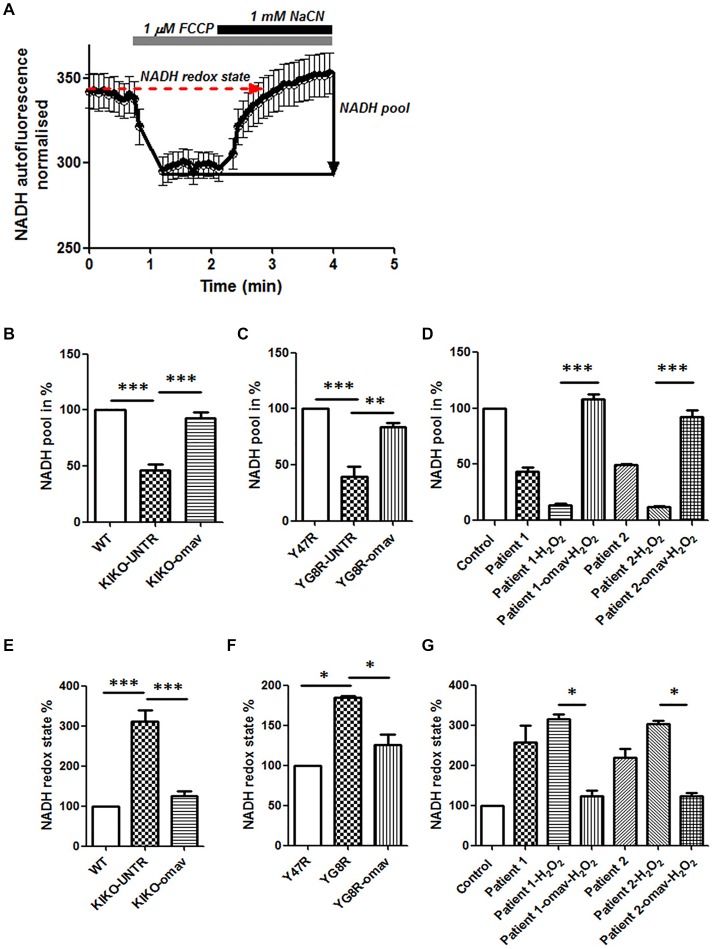
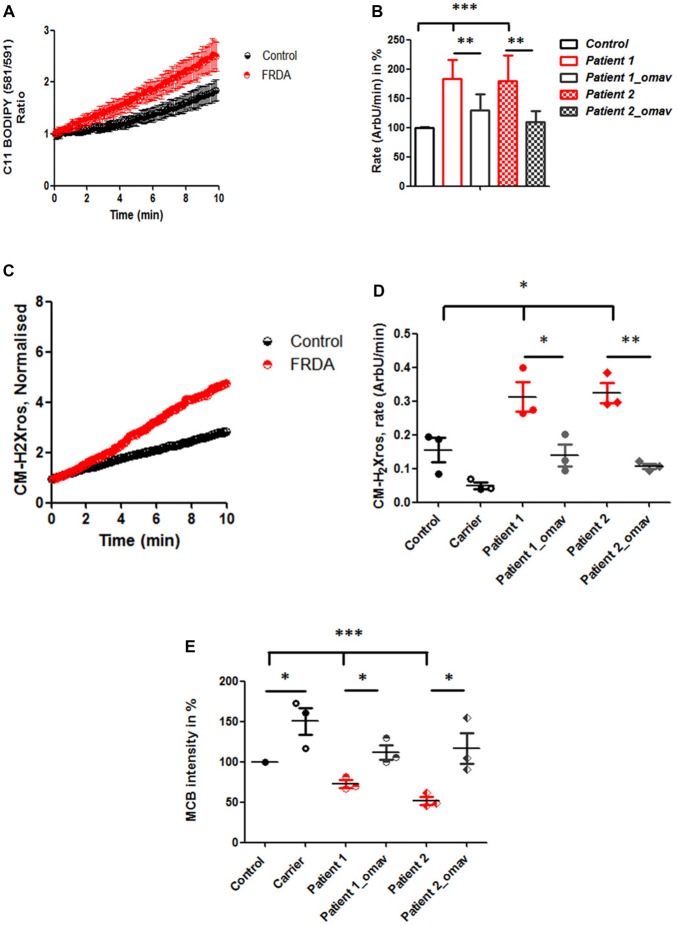
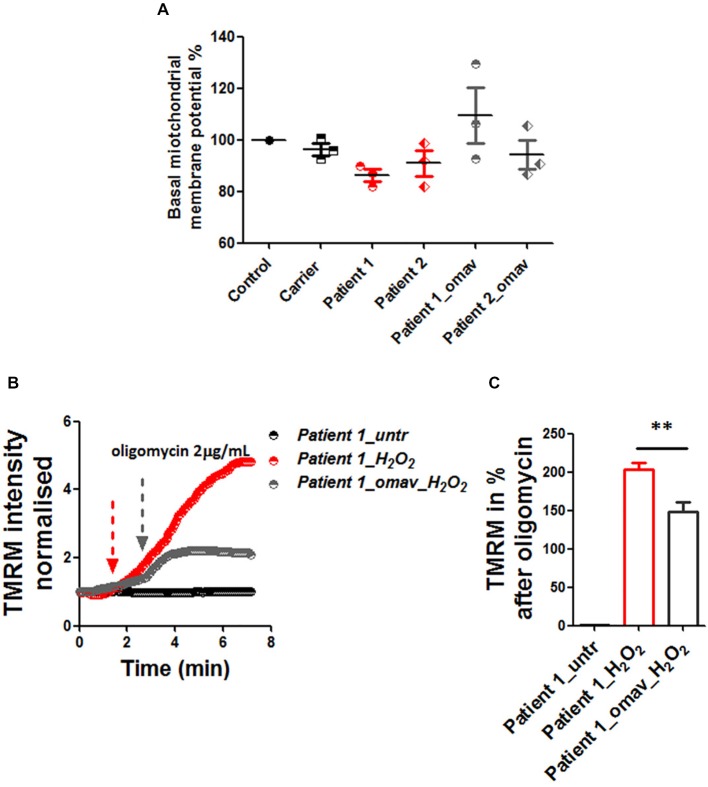
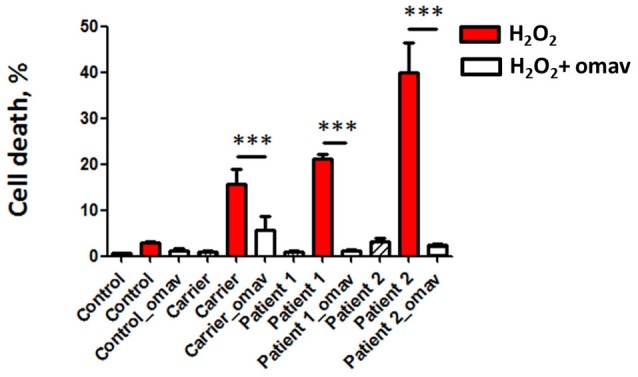
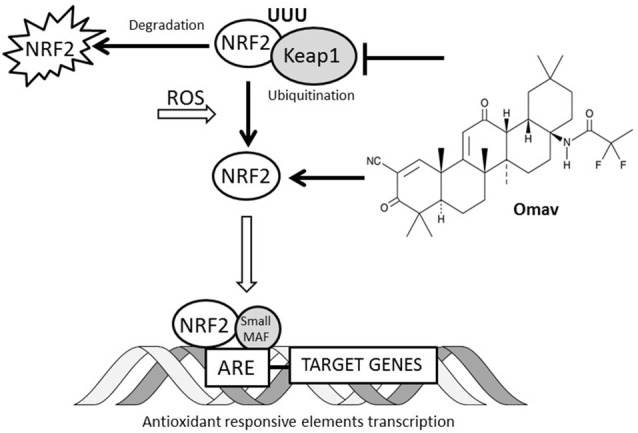
Friedreich's Ataxia (FRDA) is an autosomal recessive neurodegenerative disorder, affecting dorsal root ganglia (DRG), cerebellar dentate nuclei and heart. It is caused by a GAA repeat expansion mutation within the frataxin gene (FXN). This impedes FXN transcription resulting in a progressive decrease of the mitochondrial protein, frataxin. Increased oxidative stress leading to a chronic depletion of endogenous antioxidants affects the survival of the cells and causes neurodegeneration. In particular, cerebellar granule neurons (CGNs) show a significant increase of reactive oxygen species (ROS), lipid peroxidation and lower level of reduced glutathione (GSH). In FRDA, one of the major pathways of oxidant scavengers, the Nrf2 antioxidant pathway, is defective. Previous studies on FRDA-like CGNs showed that the reduced level of frataxin and the oxidative stress induce mitochondrial impairments. By triggering the Nrf2 endogenous pathway pharmacologically we determined whether this could promote mitochondrial fitness and counteract oxidative stress. In this work, we sought to investigate the beneficial effect of a promising Nrf2-inducer, omaveloxolone (omav), in CGNs from two FRDA mouse models, KIKO and YG8R, and human fibroblasts from patients. We found that CGNs from both KIKO and YG8R presented Complex I deficiency and that omav was able to restore substrate availability and Complex I activity. This was also confirmed in human primary fibroblasts from FRDA patients. Although fibroblasts are not the major tissue affected, we found that they show significant differences recapitulating the disease; this is therefore an important tool to investigate patients' pathophysiology. Interestingly, we found that patient fibroblasts had an increased level of endogenous lipid peroxidation and mitochondrial ROS (mROS), and lower GSH at rest. Omav was able to reverse this phenotype, protecting the cells against oxidative stress. By stimulating the cells with hydrogen peroxide (H2O2) and looking for potential mitochondrial pathophysiology, we found that fibroblasts could not maintain their mitochondrial membrane potential (ΔΨm). Remarkably, omav was protective to mitochondrial depolarization, promoting mitochondrial respiration and preventing cell death. Our results show that omav promotes Complex I activity and protect cells from oxidative stress. Omav could, therefore, be used as a novel therapeutic drug to ameliorate the pathophysiology of FRDA.
Keywords: Friedreich’s ataxia; human fibroblasts; lipid peroxidation; mitochondrial dysfunction; nuclear factor (erythroid-derived 2)-like 2; reactive oxygen species.
Figures
References
-
- Al-Mahdawi S., Pinto R. M., Varshney D., Lawrence L., Lowrie M. B., Hughes S., et al. . (2006). GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 88, 580–590. 10.1016/j.ygeno.2006.06.015 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous