

Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell-Cell Adhesion Structures
- PMID: 30067491
- PMCID: PMC6074050
- DOI: 10.1016/j.jacep.2017.12.003
Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell-Cell Adhesion Structures
Abstract
Objectives: The purpose of this study was to assess the phenotype of Filamin C (FLNC) truncating variants in dilated cardiomyopathy (DCM) and understand the mechanism leading to an arrhythmogenic phenotype.
Background: Mutations in FLNC are known to lead to skeletal myopathies, which may have an associated cardiac component. Recently, the clinical spectrum of FLNC mutations has been recognized to include a cardiac-restricted presentation in the absence of skeletal muscle involvement.
Methods: A population of 319 U.S. and European DCM cardiomyopathy families was evaluated using whole-exome and targeted next-generation sequencing. FLNC truncation probands were identified and evaluated by clinical examination, histology, transmission electron microscopy, and immunohistochemistry.
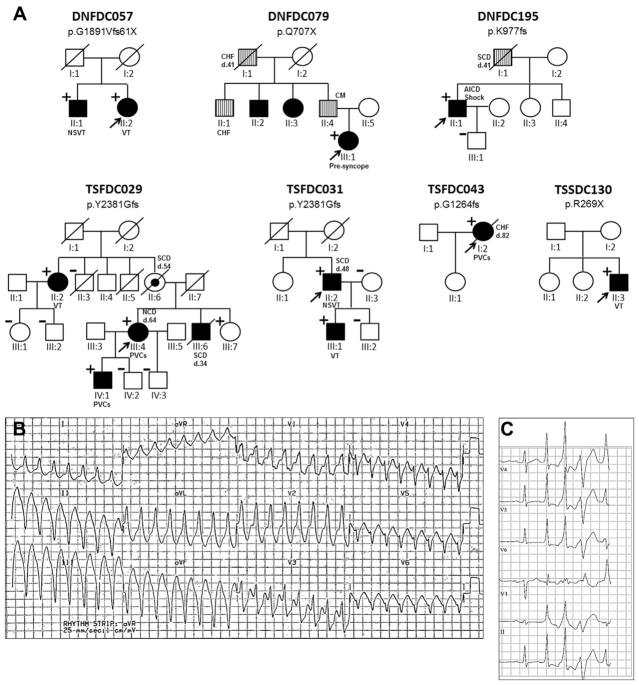
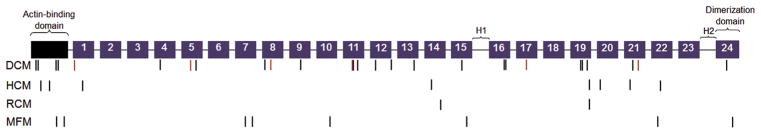
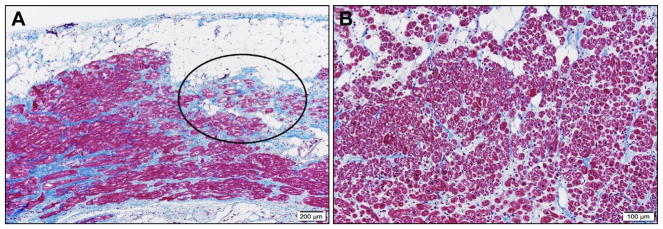
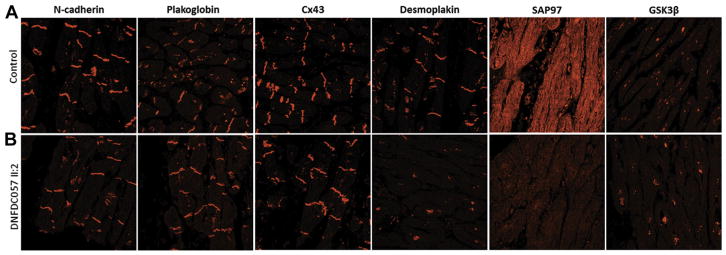
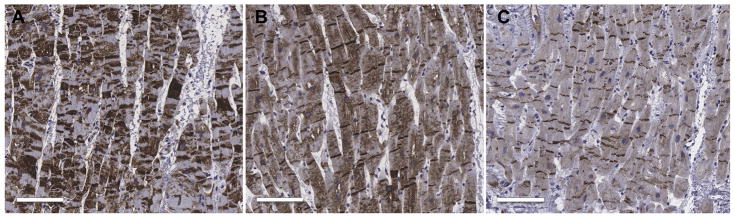
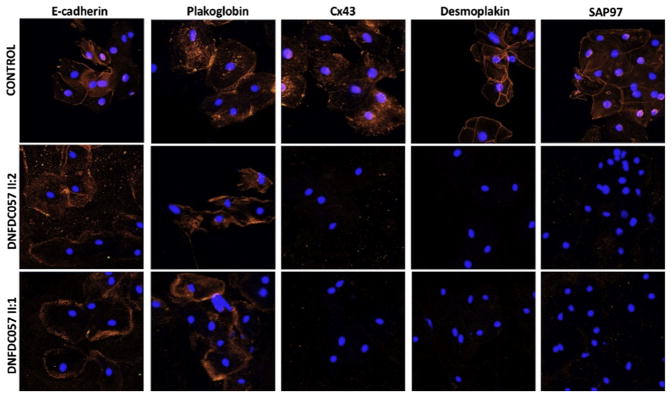
Results: A total of 13 individuals in 7 families (2.2%) were found to harbor 6 different FLNC truncation variants (2 stopgain, 1 frameshift, and 3 splicing). Of the 13 FLNC truncation carriers, 11 (85%) had either ventricular arrhythmias or sudden cardiac death, and 5 (38%) presented with evidence of right ventricular dilation. Pathology analysis of 2 explanted hearts from affected FLNC truncation carriers showed interstitial fibrosis in the right ventricle and epicardial fibrofatty infiltration in the left ventricle. Ultrastructural findings included occasional disarray of Z-discs within the sarcomere. Immunohistochemistry showed normal plakoglobin signal at cell-cell junctions, but decreased signals for desmoplakin and synapse-associated protein 97 in the myocardium and buccal mucosa.
Conclusions: We found FLNC truncating variants, present in 2.2% of DCM families, to be associated with a cardiac-restricted arrhythmogenic DCM phenotype characterized by a high risk of life-threatening ventricular arrhythmias and a pathological cellular phenotype partially overlapping with arrhythmogenic right ventricular cardiomyopathy.
Keywords: Filamin C; arrhythmias; arrhythmogenic dilated cardiomyopathy; cardiovascular genetics; familial dilated cardiomyopathy; heart failure.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

Comment in
-
Filamin C: A New Arrhythmogenic Cardiomyopathy-Causing Gene?JACC Clin Electrophysiol. 2018 Apr;4(4):515-517. doi: 10.1016/j.jacep.2018.01.004. JACC Clin Electrophysiol. 2018. PMID: 30067492 No abstract available.
References
-
- Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36(18):1123–35a. - PubMed
-
- Daughenbaugh LA. Cardiomyopathy: an overview. J Nurse Pract. 2007;3:248–58.
-
- Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531–47. - PubMed
-
- Gontier Y, Taivainen A, Fontao L, et al. The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sarcolemma via muscle-specific filamins. J Cell Sci. 2005;118(pt 16):3739–49. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
