

First principles-based multiscale atomistic methods for input into first principles nonequilibrium transport across interfaces
- PMID: 30076227
- PMCID: PMC6744898
- DOI: 10.1073/pnas.1800035115
First principles-based multiscale atomistic methods for input into first principles nonequilibrium transport across interfaces
Abstract
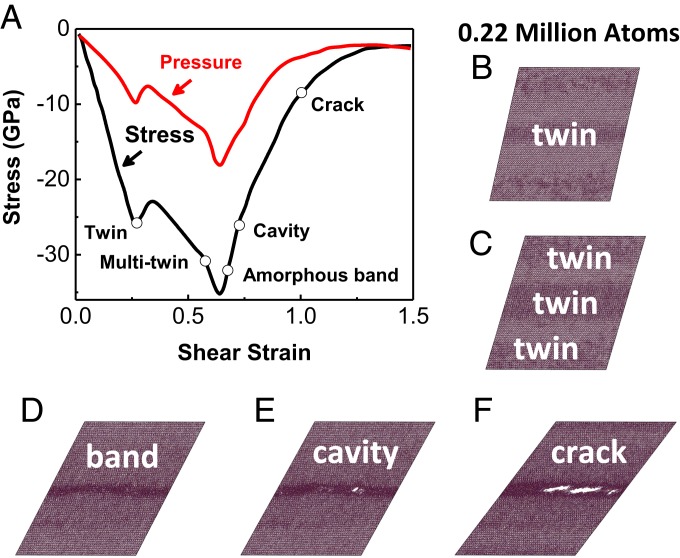
This issue of PNAS features "nonequilibrium transport and mixing across interfaces," with several papers describing the nonequilibrium coupling of transport at interfaces, including mesoscopic and macroscopic dynamics in fluids, plasma, and other materials over scales from microscale to celestial. Most such descriptions describe the materials in terms of the density and equations of state rather than specific atomic structures and chemical processes. It is at interfacial boundaries where such atomistic information is most relevant. However, there is not yet a practical way to couple these phenomena with the atomistic description of chemistry. The starting point for including such information is the quantum mechanics (QM). However, practical QM calculations are limited to a hundred atoms for dozens of picoseconds, far from the scales required to inform the continuum level with the proper atomistic description. To bridge this enormous gap, we need to develop practical methods to extend the scale of the atomistic simulation by several orders of magnitude while retaining the level of QM accuracy in describing the chemical process. These developments would enable continuum modeling of turbulent transport at interfaces to incorporate the relevant chemistry. In this perspective, we will focus on recent progress in accomplishing these extensions in first principles-based atomistic simulations and the strategies being pursued to increase the accuracy of very large scales while dramatically decreasing the computational effort.
Keywords: electron force field; molecular dynamics; multiscale simulation; quantum mechanics; reactive force fields.
Conflict of interest statement
Author contributions: W.A.G. designed research; T.C., A.J.-B., Q.A., D.V.I., and S.N. performed research; and T.C., A.J.-B., Q.A., D.V.I., S.N., and W.A.G. wrote the paper. The authors declare no conflict of interest.
Figures









References
-
- Chenoweth K, van Duin ACT, Goddard WA. Reaxff reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J Phys Chem A. 2008;112:1040–1053. - PubMed
-
- van Duin ACT, Dasgupta S, Lorant F, Goddard WA. Reaxff: A reactive force field for hydrocarbons. J Phys Chem A. 2011;105:9396–9409.
-
- Rappe AK, Goddard WA. Charge equilibration for molecular dynamics simulations. J Phys Chem. 1991;95:3358–3363.
-
- Naserifar S, Brooks DJ, Goddard WA, Cvicek V. Polarizable charge equilibration model for predicting accurate electrostatic interactions in molecules and solids. J Chem Phys. 2017;146:124117. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
