

A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility
- PMID: 30078704
- PMCID: PMC6158300
- DOI: 10.1016/j.cell.2018.06.052
A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility
Abstract
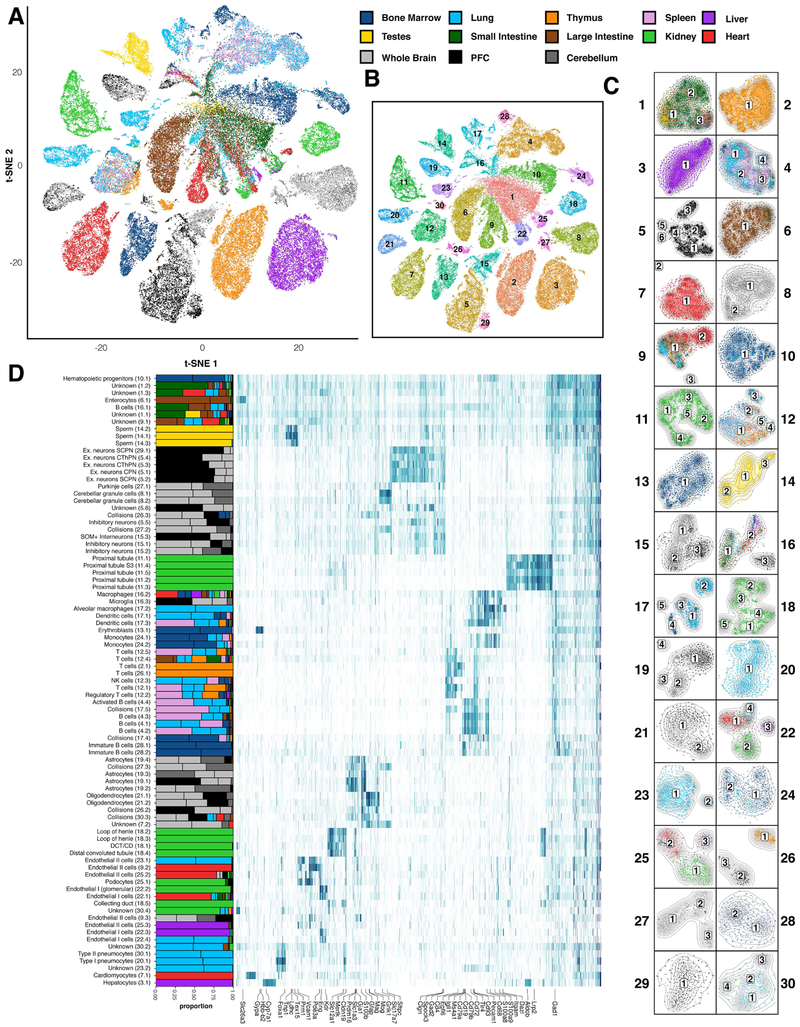
We applied a combinatorial indexing assay, sci-ATAC-seq, to profile genome-wide chromatin accessibility in ∼100,000 single cells from 13 adult mouse tissues. We identify 85 distinct patterns of chromatin accessibility, most of which can be assigned to cell types, and ∼400,000 differentially accessible elements. We use these data to link regulatory elements to their target genes, to define the transcription factor grammar specifying each cell type, and to discover in vivo correlates of heterogeneity in accessibility within cell types. We develop a technique for mapping single cell gene expression data to single-cell chromatin accessibility data, facilitating the comparison of atlases. By intersecting mouse chromatin accessibility with human genome-wide association summary statistics, we identify cell-type-specific enrichments of the heritability signal for hundreds of complex traits. These data define the in vivo landscape of the regulatory genome for common mammalian cell types at single-cell resolution.
Keywords: ATAC-seq; GWAS; chromatin; chromatin accessibility; epigenetics; epigenomics; regulatory; single cell.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures






References
-
- Arendt D, Musser JM, Baker CVH, Bergman A, Cepko C, Erwin DH, Pavlicev M, Schlosser G, Widder S, Laubichler MD, et al. (2016). The origin and evolution of cell types. Nat. Rev. Genet 17, 744–757. - PubMed
-
- Bello SM, and Eppig JT (2016). Inferring gene-to-phenotype and gene-to-disease relationships at Mouse Genome Informatics: challenges and solutions. J. Biomed. Semantics 7, 14.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
