

Idiopathic Pulmonary Fibrosis (IPF): An Overview
- PMID: 30082599
- PMCID: PMC6111543
- DOI: 10.3390/jcm7080201
Idiopathic Pulmonary Fibrosis (IPF): An Overview
Abstract
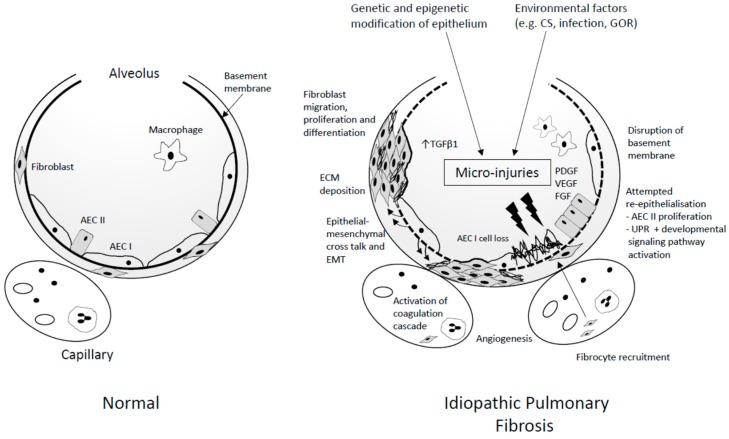
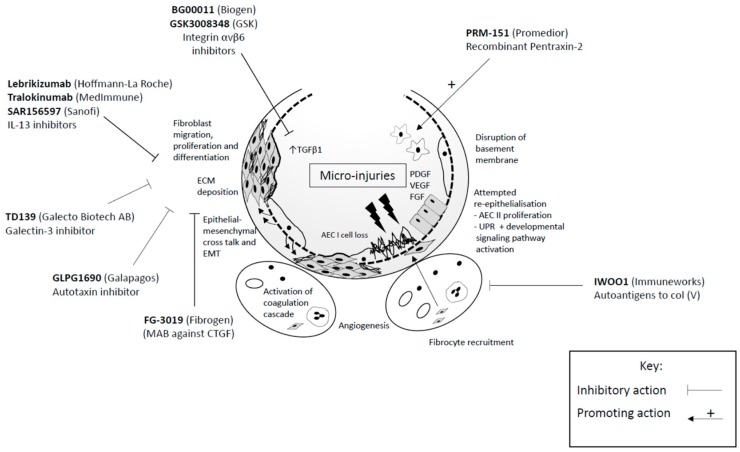
Idiopathic pulmonary fibrosis (IPF) is an interstitial lung disease characterised by chronic, progressive scarring of the lungs and the pathological hallmark of usual interstitial pneumonia. Current paradigms suggest alveolar epithelial cell damage is a key initiating factor. Globally, incidence of the disease is rising, with associated high morbidity, mortality, and economic healthcare burden. Diagnosis relies on a multidisciplinary team approach with exclusion of other causes of interstitial lung disease. Over recent years, two novel antifibrotic therapies, pirfenidone and nintedanib, have been developed, providing treatment options for many patients with IPF, with several other agents in early clinical trials. Current efforts are directed at identifying key biomarkers that may direct more customized patient-centred healthcare to improve outcomes for these patients in the future.
Keywords: idiopathic pulmonary fibrosis; interstitial lung disease; nintedanib; pirfenidone.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
