

SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions
- PMID: 30085201
- PMCID: PMC6237738
- DOI: 10.1093/nar/gky691
SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions
Abstract
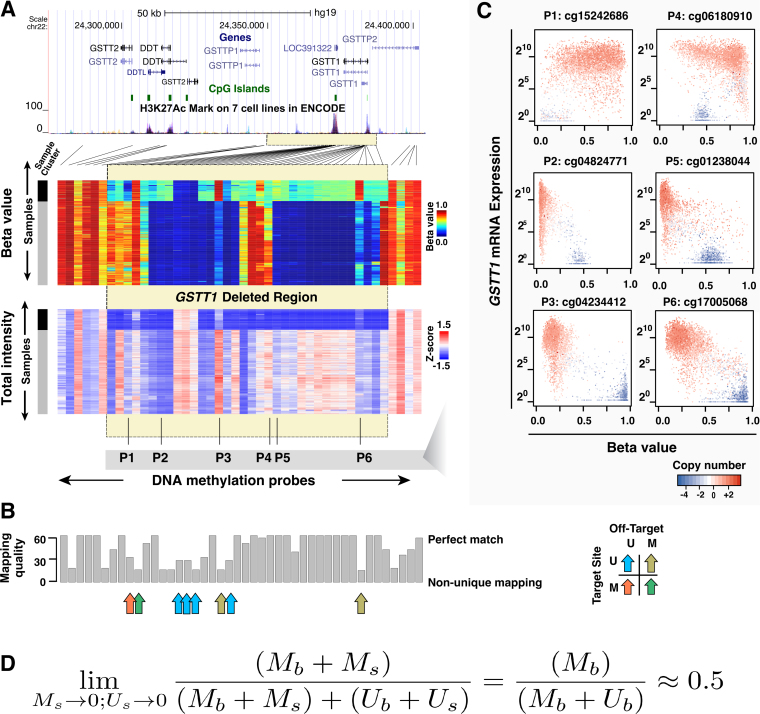
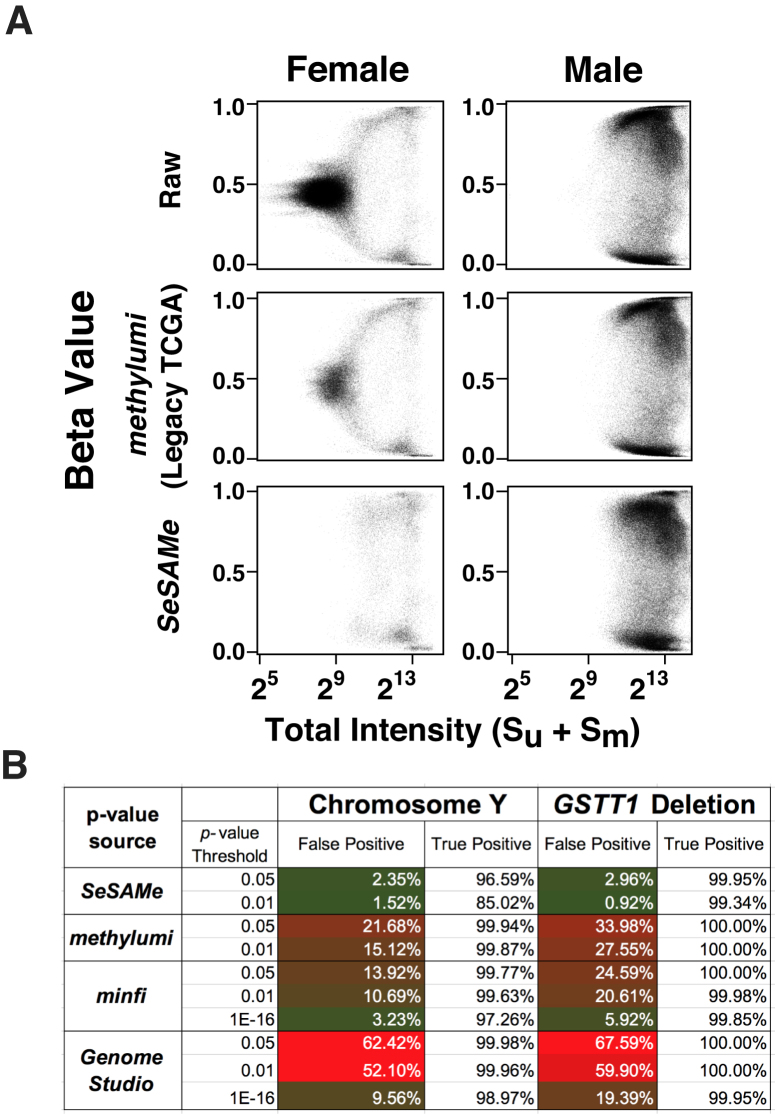
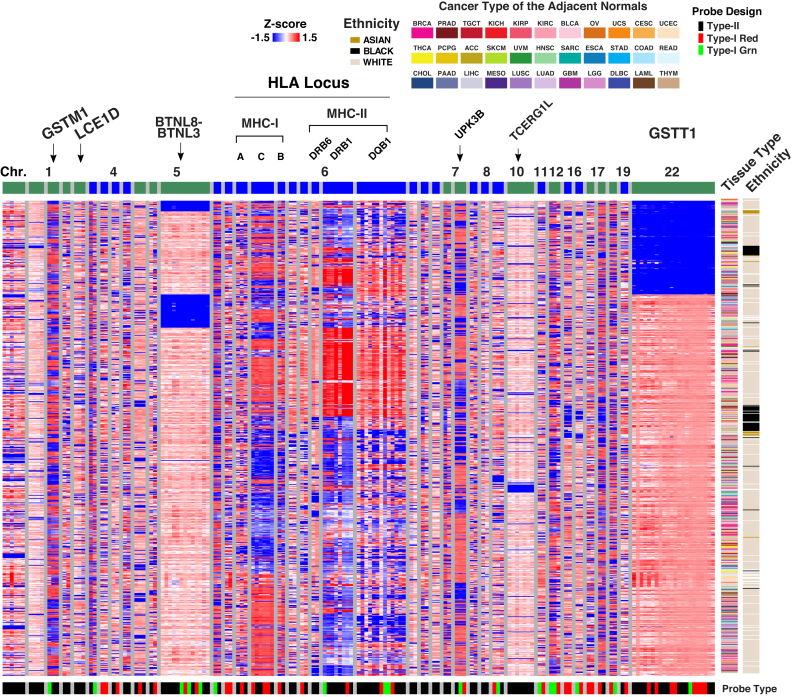
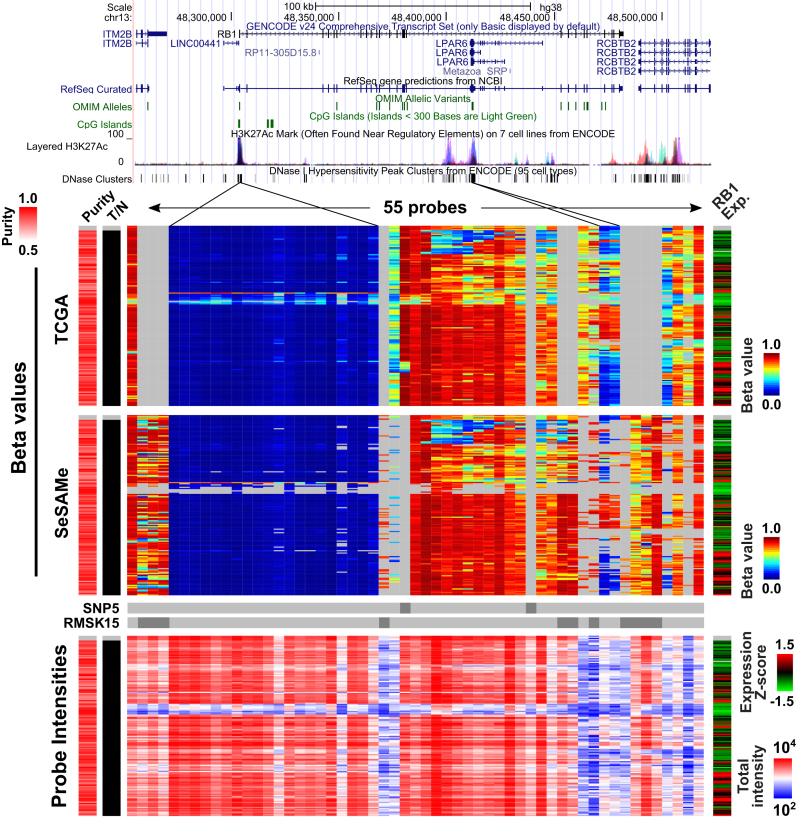
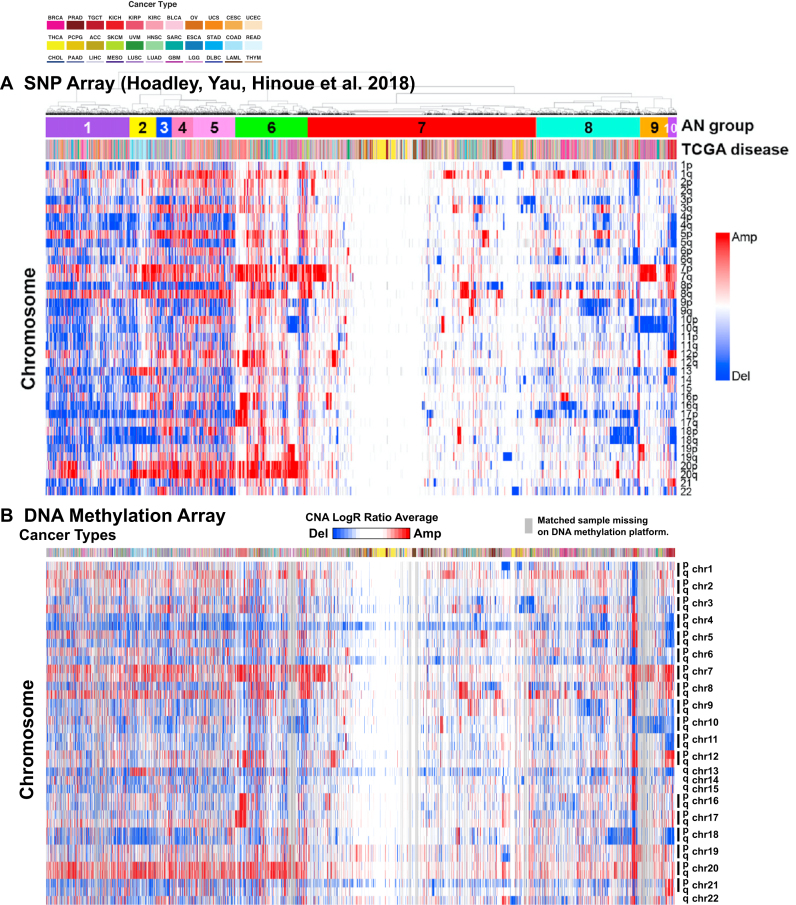
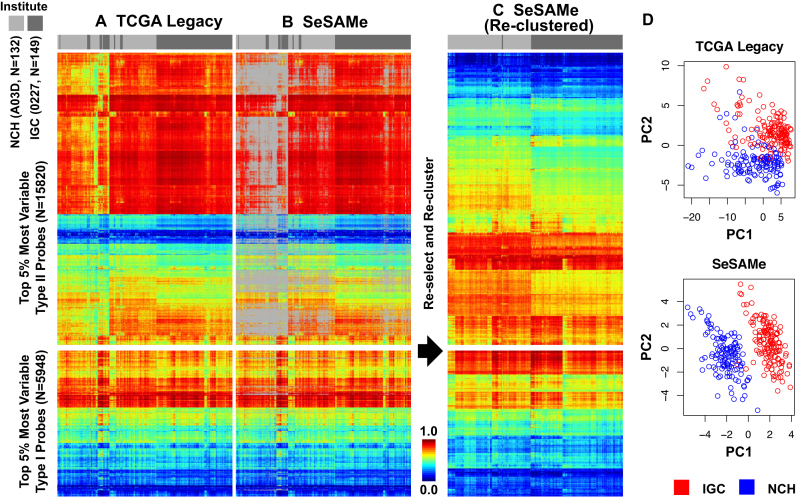
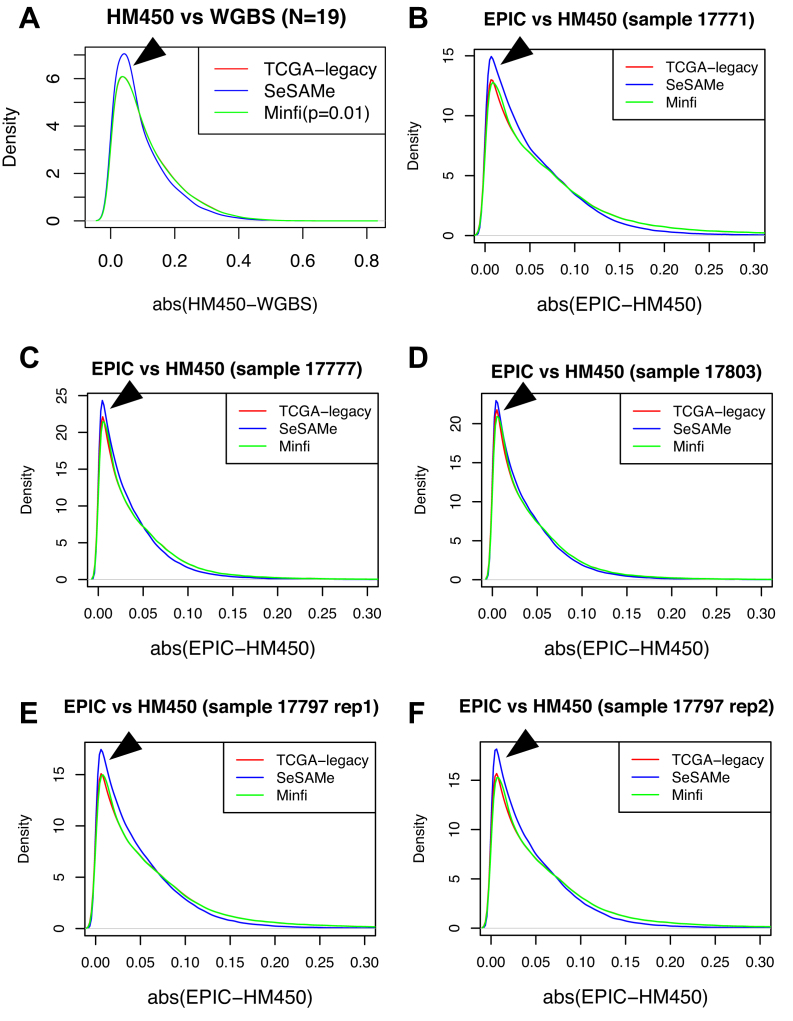
We report a new class of artifacts in DNA methylation measurements from Illumina HumanMethylation450 and MethylationEPIC arrays. These artifacts reflect failed hybridization to target DNA, often due to germline or somatic deletions and manifest as incorrectly reported intermediate methylation. The artifacts often survive existing preprocessing pipelines, masquerade as epigenetic alterations and can confound discoveries in epigenome-wide association studies and studies of methylation-quantitative trait loci. We implement a solution, P-value with out-of-band (OOB) array hybridization (pOOBAH), in the R package SeSAMe. Our method effectively masks deleted and hyperpolymorphic regions, reducing or eliminating spurious reports of epigenetic silencing at oft-deleted tumor suppressor genes such as CDKN2A and RB1 in cases with somatic deletions. Furthermore, our method substantially decreases technical variation whilst retaining biological variation, both within and across HM450 and EPIC platform measurements. SeSAMe provides a light-weight, modular DNA methylation data analysis suite, with a performant implementation suitable for efficient analysis of thousands of samples.
Figures

References
-
- Teschendorff A.E., Relton C.L.. Statistical and integrative system-level analysis of DNA methylation data. Nat. Rev. Genet. 2018; 19:129–147. - PubMed
-
- Flanagan J.M. Epigenome-wide association studies (EWAS): past, present, and future. Methods Mol. Biol. 2015; 1238:51–63. - PubMed
-
- van Dijk S.J., Peters T.J., Buckley M., Zhou J., Jones P.A., Gibson R.A., Makrides M., Muhlhausler B.S., Molloy P.L.. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int. J. Obes. 2018; 42:28–35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
