

Pathophysiology and treatment of cerebral edema in traumatic brain injury
- PMID: 30086289
- PMCID: PMC6309515
- DOI: 10.1016/j.neuropharm.2018.08.004
Pathophysiology and treatment of cerebral edema in traumatic brain injury
Abstract
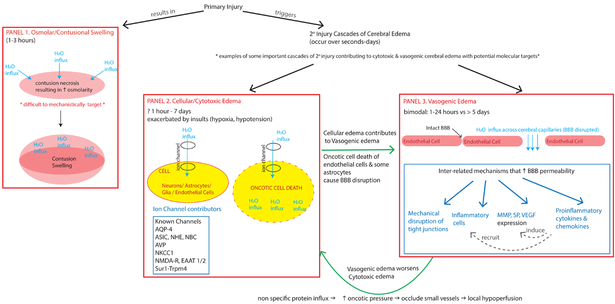
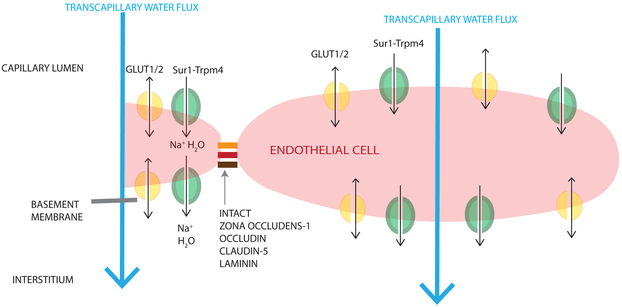
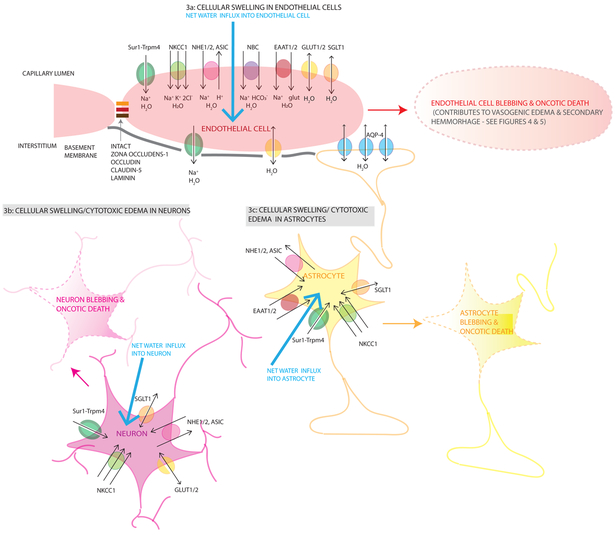
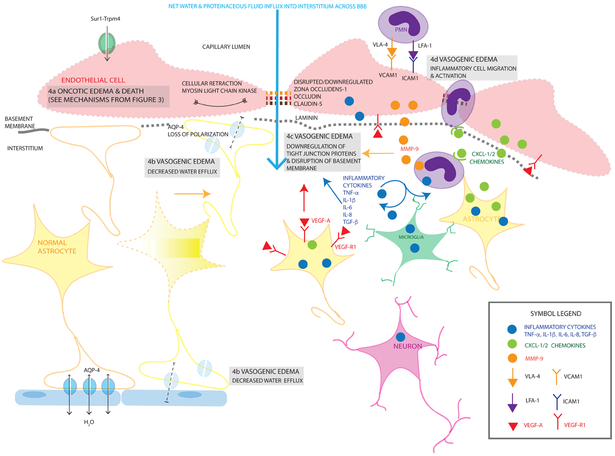
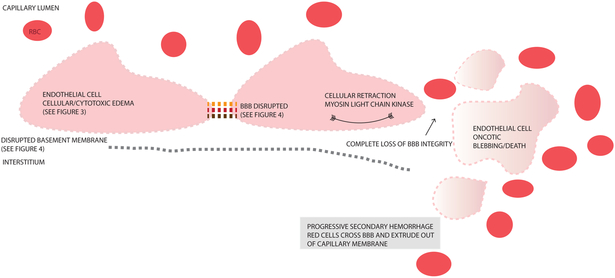
Cerebral edema (CE) and resultant intracranial hypertension are associated with unfavorable prognosis in traumatic brain injury (TBI). CE is a leading cause of in-hospital mortality, occurring in >60% of patients with mass lesions, and ∼15% of those with normal initial computed tomography scans. After treatment of mass lesions in severe TBI, an important focus of acute neurocritical care is evaluating and managing the secondary injury process of CE and resultant intracranial hypertension. This review focuses on a contemporary understanding of various pathophysiologic pathways contributing to CE, with a subsequent description of potential targeted therapies. There is a discussion of identified cellular/cytotoxic contributors to CE, as well as mechanisms that influence blood-brain-barrier (BBB) disruption/vasogenic edema, with the caveat that this distinction may be somewhat artificial since molecular processes contributing to these pathways are interrelated. While an exhaustive discussion of all pathways with putative contributions to CE is beyond the scope of this review, the roles of some key contributors are highlighted, and references are provided for further details. Potential future molecular targets for treating CE are presented based on pathophysiologic mechanisms. We thus aim to provide a translational synopsis of present and future strategies targeting CE after TBI in the context of a paradigm shift towards precision medicine. This article is part of the Special Issue entitled "Novel Treatments for Traumatic Brain Injury".
Keywords: Cerebral edema; Cytotoxic edema; Ionic edema; Traumatic brain injury; Vasogenic edema.
Copyright © 2018 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures
References
-
- Allen CJ, Subhawong TK, Hanna MM, Chelala L, Bullock MR, Schulman CI, Proctor KG, 2018. Does Vasopressin Exacerbate Cerebral Edema in Patients with Severe Traumatic Brain Injury? Am. Surg 84, 43–50. - PubMed
-
- Alluri H, Wilson RL, Anasooya Shaji C, Wiggins-Dohlvik K, Patel S, Liu Y, Peng X, Beeram MR, Davis ML, Huang JH, Tharakan B, 2016. Melatonin Preserves Blood-Brain Barrier Integrity and Permeability via Matrix Metalloproteinase-9 Inhibition. PLoS One 11, e0154427. doi:10.1371/journal.pone.0154427 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
