

Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation
- PMID: 30087121
- PMCID: PMC6153299
- DOI: 10.1074/jbc.RA117.001469
Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation
Abstract
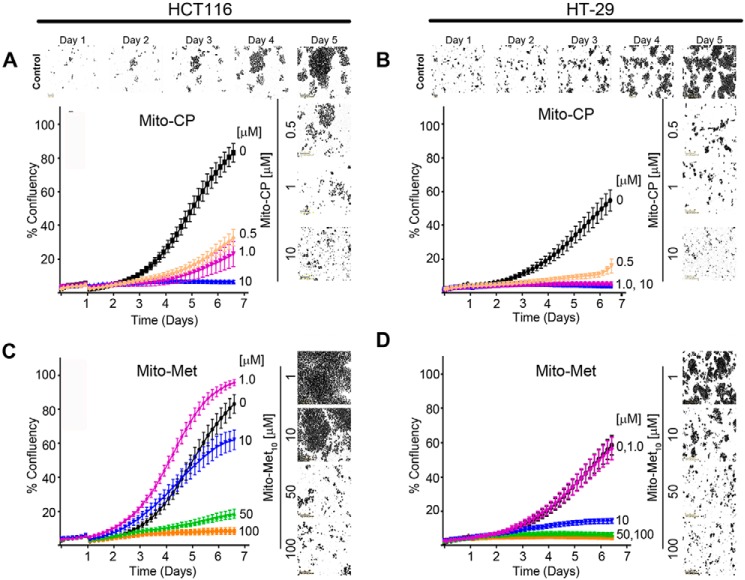
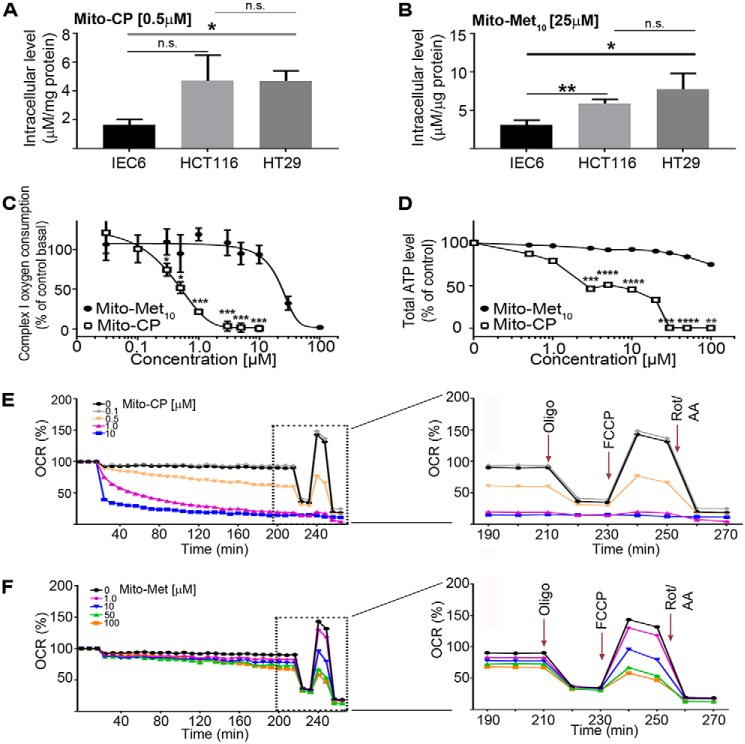
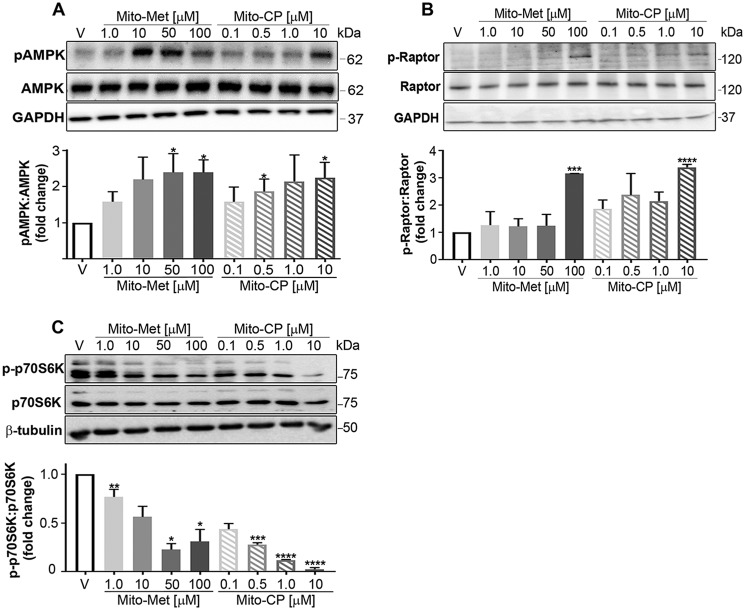
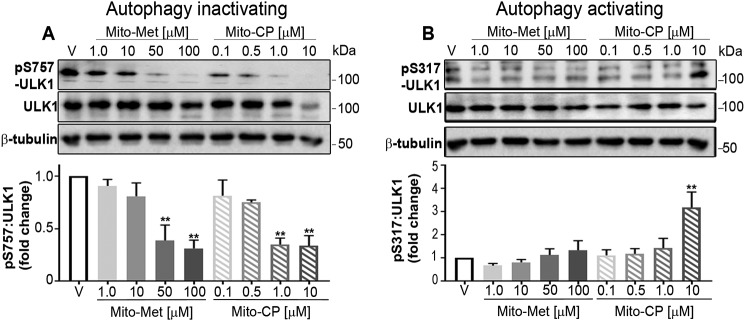
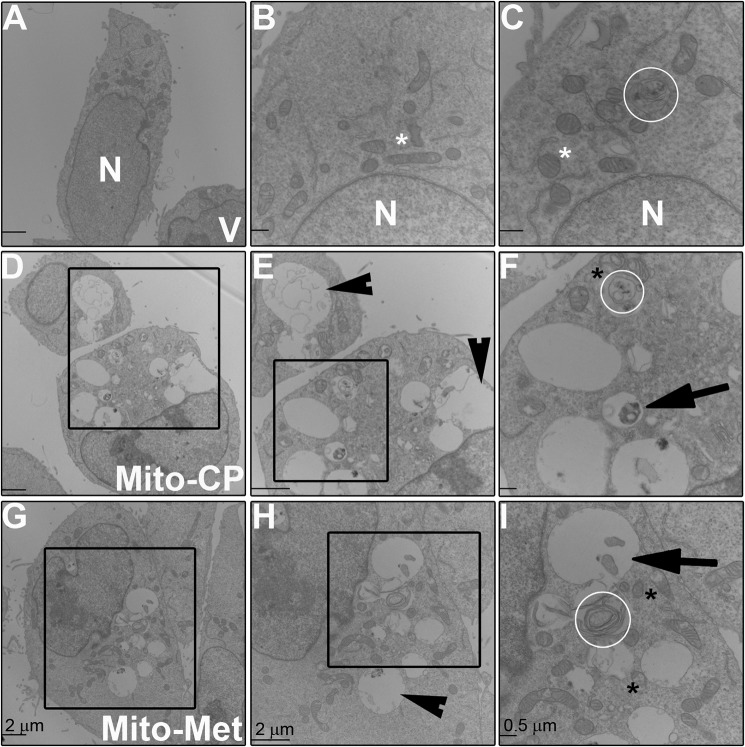
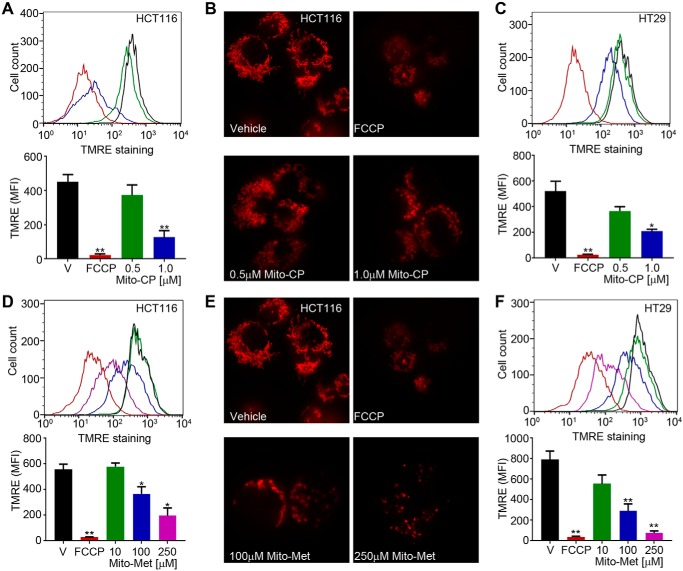
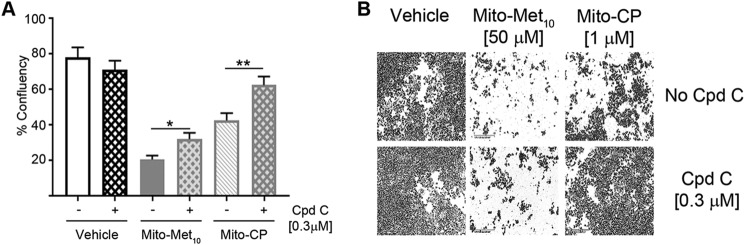
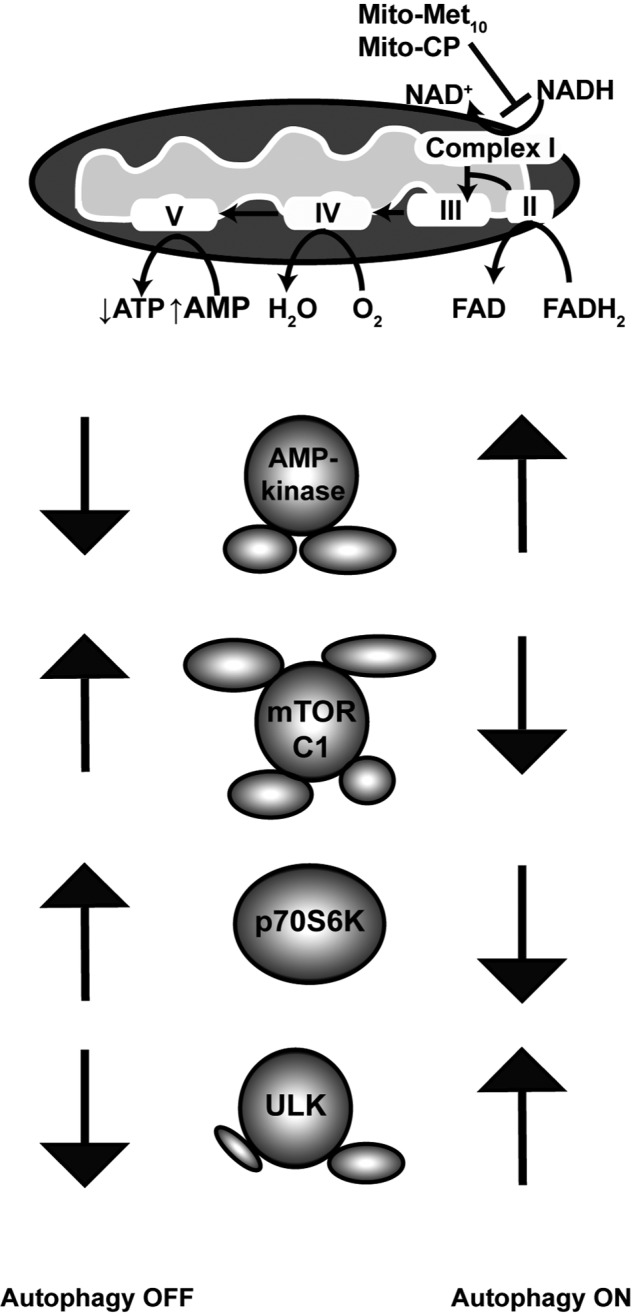
Mutations in the KRAS proto-oncogene are present in 50% of all colorectal cancers and are increasingly associated with chemotherapeutic resistance to frontline biologic drugs. Accumulating evidence indicates key roles for overactive KRAS mutations in the metabolic reprogramming from oxidative phosphorylation to aerobic glycolysis in cancer cells. Here, we sought to exploit the more negative membrane potential of cancer cell mitochondria as an untapped avenue for interfering with energy metabolism in KRAS variant-containing and KRAS WT colorectal cancer cells. Mitochondrial function, intracellular ATP levels, cellular uptake, energy sensor signaling, and functional effects on cancer cell proliferation were assayed. 3-Carboxyl proxyl nitroxide (Mito-CP) and Mito-Metformin, two mitochondria-targeted compounds, depleted intracellular ATP levels and persistently inhibited ATP-linked oxygen consumption in both KRAS WT and KRAS variant-containing colon cancer cells and had only limited effects on nontransformed intestinal epithelial cells. These anti-proliferative effects reflected the activation of AMP-activated protein kinase (AMPK) and the phosphorylation-mediated suppression of the mTOR target ribosomal protein S6 kinase B1 (RPS6KB1 or p70S6K). Moreover, Mito-CP and Mito-Metformin released Unc-51-like autophagy-activating kinase 1 (ULK1) from mTOR-mediated inhibition, affected mitochondrial morphology, and decreased mitochondrial membrane potential, all indicators of mitophagy. Pharmacological inhibition of the AMPK signaling cascade mitigated the anti-proliferative effects of Mito-CP and Mito-Metformin. This is the first demonstration that drugs selectively targeting mitochondria induce mitophagy in cancer cells. Targeting bioenergetic metabolism with mitochondria-targeted drugs to stimulate mitophagy provides an attractive approach for therapeutic intervention in KRAS WT and overactive mutant-expressing colon cancer.
Keywords: AMP-activated kinase (AMPK); KRAS proto-oncogene; cancer biology; cell growth; cell proliferation; colon cancer; metformin; mitochondrial metabolism; mitophagy.
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
M. B. D. has ownership interests in Protein Foundry, LLC
Figures
References
-
- American Cancer Society. Cancer Facts & Figures 2017. American Cancer Society, Atlanta, GA
-
- De Roock W., Claes B., Bernasconi D., De Schutter J., Biesmans B., Fountzilas G., Kalogeras K. T., Kotoula V., Papamichael D., Laurent-Puig P., Penault-Llorca F., Rougier P., Vincenzi B., Santini D., Tonini G., et al. (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 11, 753–762 10.1016/S1470-2045(10)70130-3 - DOI - PubMed
-
- Misale S., Yaeger R., Hobor S., Scala E., Janakiraman M., Liska D., Valtorta E., Schiavo R., Buscarino M., Siravegna G., Bencardino K., Cercek A., Chen C. T., Veronese S., Zanon C., et al. (2012) Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486, 532–536 10.1038/nature11156 - DOI - PMC - PubMed
-
- Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G. M., Budinger G. R., and Chandel N. S. (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 8788–8793 10.1073/pnas.1003428107 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
