

A Comparative Study of Modern Homology Modeling Algorithms for Rhodopsin Structure Prediction
- PMID: 30087916
- PMCID: PMC6068592
- DOI: 10.1021/acsomega.8b00721
A Comparative Study of Modern Homology Modeling Algorithms for Rhodopsin Structure Prediction
Abstract
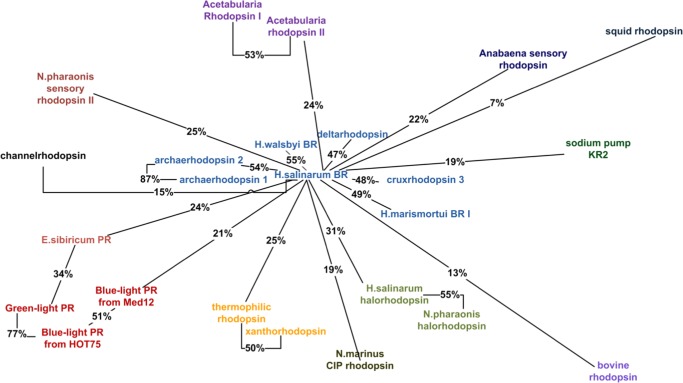
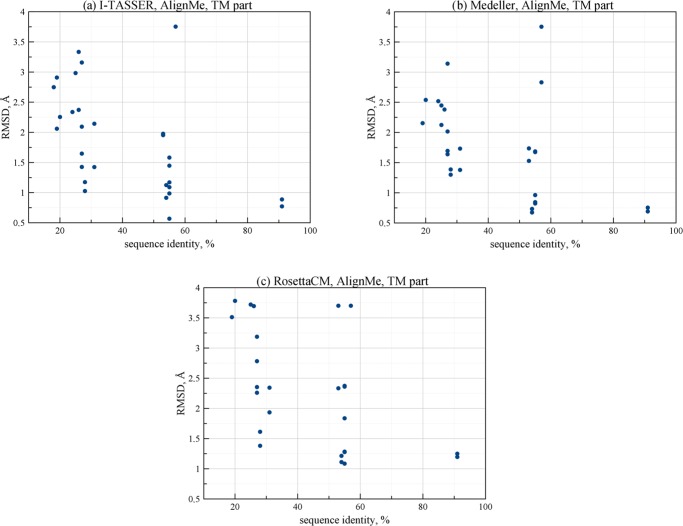
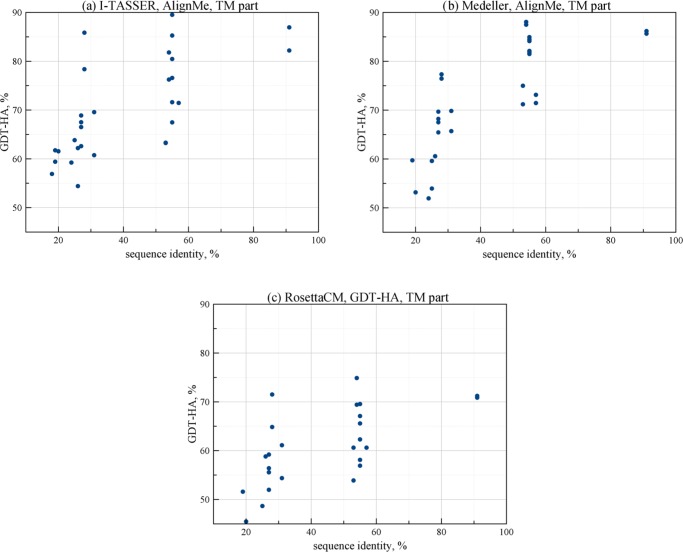
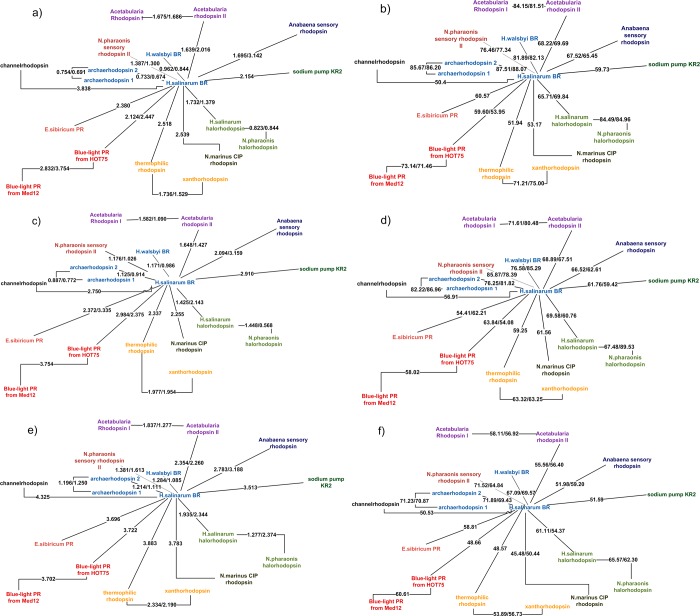
Rhodopsins are seven α-helical membrane proteins that are of great importance in chemistry, biology, and modern biotechnology. Any in silico study on rhodopsin properties and functioning requires a high-quality three-dimensional structure. Due to particular difficulties with obtaining membrane protein structures from the experiment, in silico prediction of the three-dimensional rhodopsin structure based only on its primary sequence is an especially important task. For the last few years, significant progress was made in the field of protein structure prediction, especially for methods based on comparative modeling. However, the majority of this progress was made for soluble proteins and further investigations are needed to achieve similar progress for membrane proteins. In this paper, we evaluate the performance of modern protein structure prediction methodologies (implemented in the Medeller, I-TASSER, and Rosetta packages) for their ability to predict rhodopsin structures. Three widely used methodologies were considered: two general methodologies that are commonly applied to soluble proteins and a methodology that uses constraints that are specific for membrane proteins. The test pool consisted of 36 target-template pairs with different sequence similarities that was constructed on the basis of 24 experimental rhodopsin structures taken from the RCSB database. As a result, we showed that all three considered methodologies allow obtaining rhodopsin structures with the quality that is close to the crystallographic one (root mean square deviation (RMSD) of the predicted structure from the corresponding X-ray structure up to 1.5 Å) if the target-template sequence identity is higher than 40%. Moreover, all considered methodologies provided structures of average quality (RMSD < 4.0 Å) if the target-template sequence identity is higher than 20%. Such structures can be subsequently used for further investigation of molecular mechanisms of protein functioning and for the development of modern protein-based biotechnologies.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
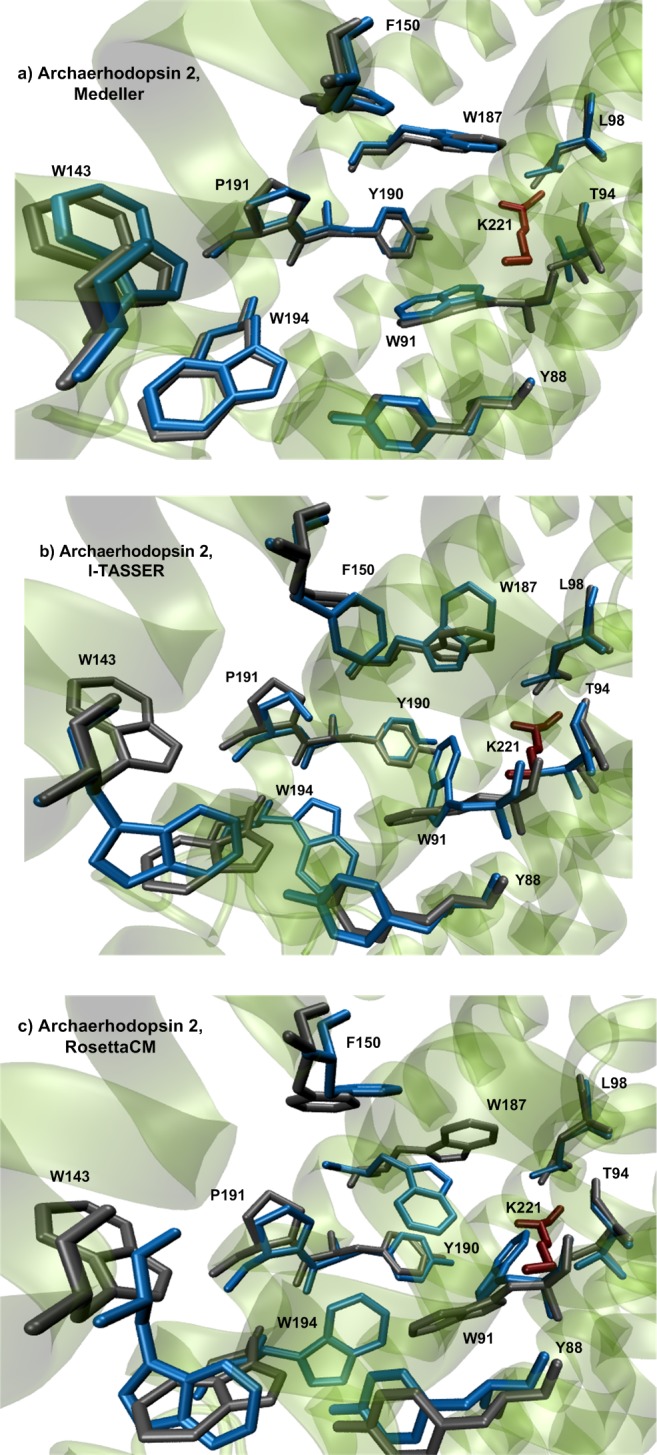
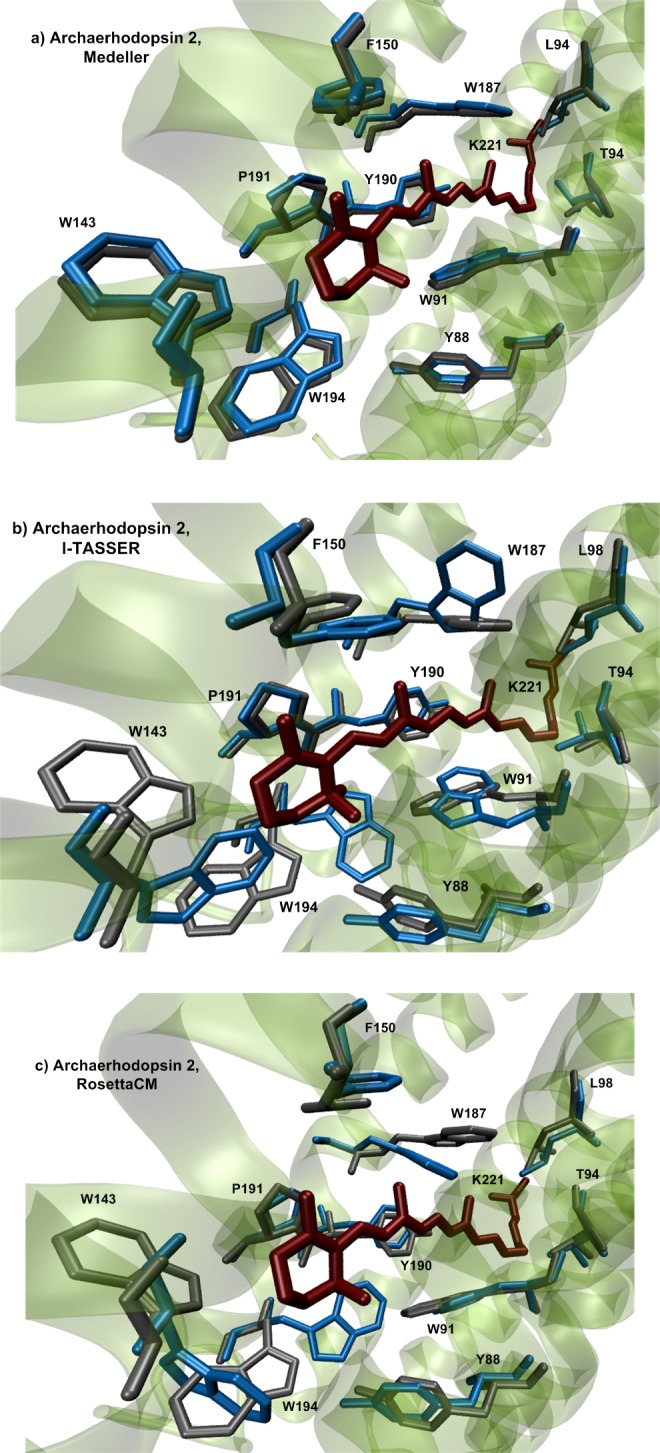
References
-
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M. S.; Eramian D.; Shen M.-y.; Pieper U.; Sali A.. Comparative Protein Structure Modeling Using MODELLER. In Current Protocols in Protein Science; John Wiley & Sons, Inc., 2001.
-
- Floudas C.; Fung H.; McAllister S.; Mönnigmann M.; Rajgaria R. Advances in protein structure prediction and de novo protein design: A review. Chem. Eng. Sci. 2006, 61, 966–988. 10.1016/j.ces.2005.04.009. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
