

PRS-on-Spark (PRSoS): a novel, efficient and flexible approach for generating polygenic risk scores
- PMID: 30089455
- PMCID: PMC6083617
- DOI: 10.1186/s12859-018-2289-9
PRS-on-Spark (PRSoS): a novel, efficient and flexible approach for generating polygenic risk scores
Abstract
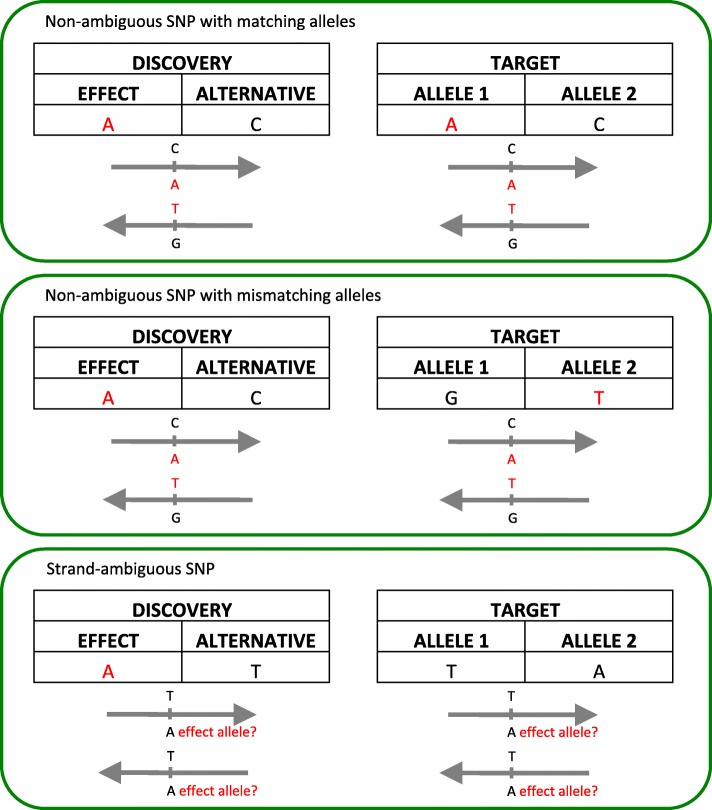
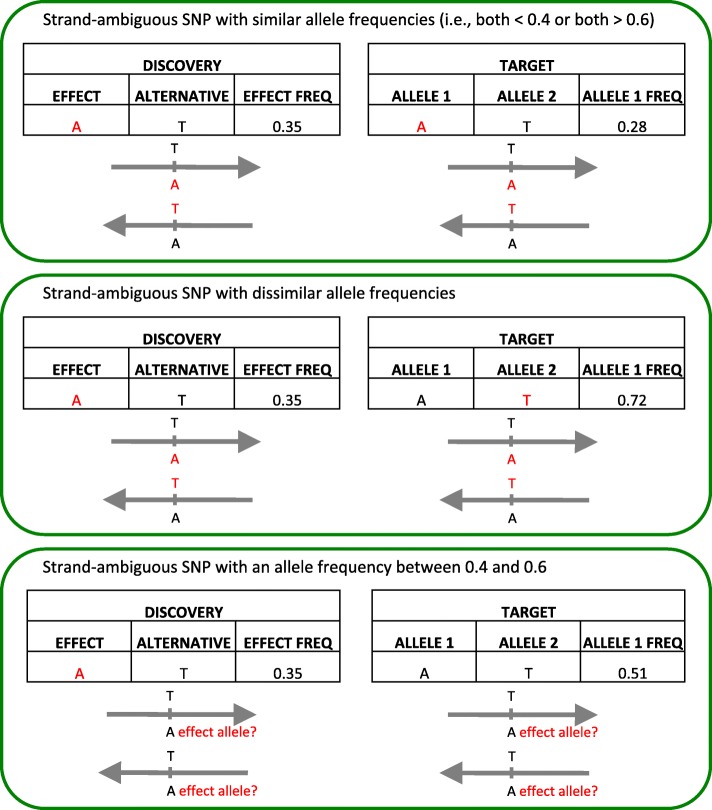
Background: Polygenic risk scores (PRS) describe the genomic contribution to complex phenotypes and consistently account for a larger proportion of variance in outcome than single nucleotide polymorphisms (SNPs) alone. However, there is little consensus on the optimal data input for generating PRS, and existing approaches largely preclude the use of imputed posterior probabilities and strand-ambiguous SNPs i.e., A/T or C/G polymorphisms. Our ability to predict complex traits that arise from the additive effects of a large number of SNPs would likely benefit from a more inclusive approach.
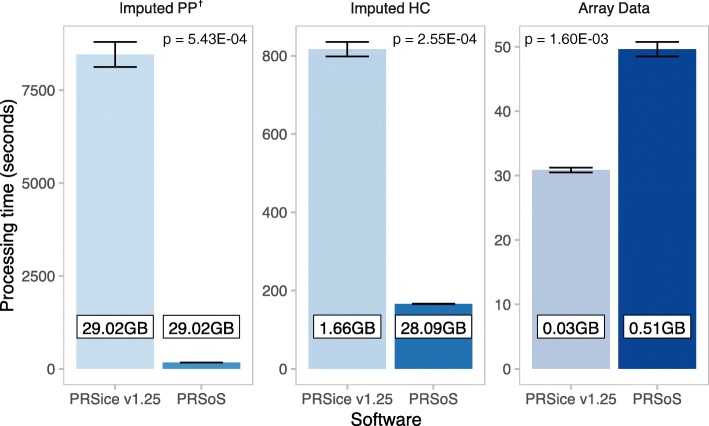
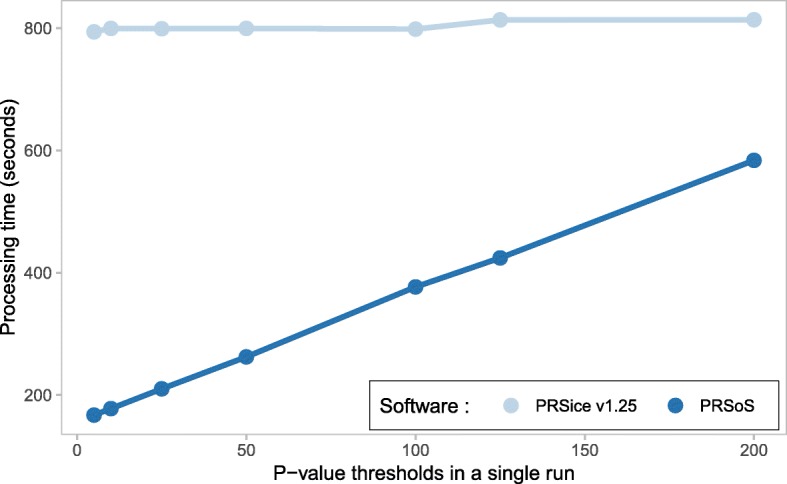
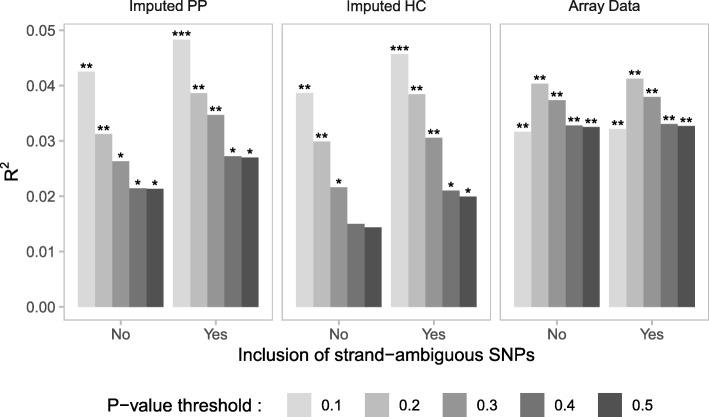
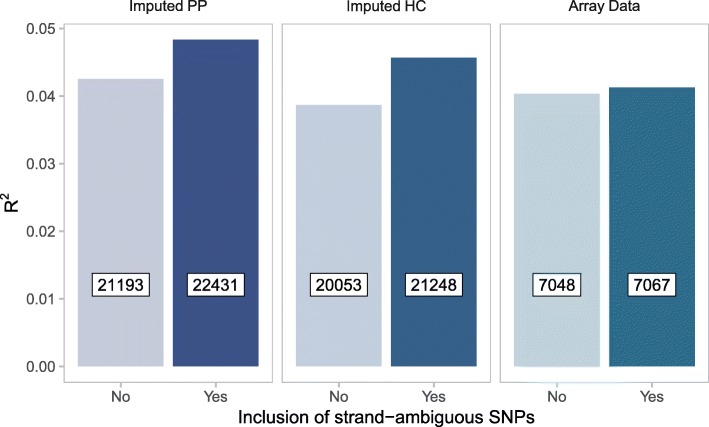
Results: We developed PRS-on-Spark (PRSoS), a software implemented in Apache Spark and Python that accommodates different data inputs and strand-ambiguous SNPs to calculate PRS. We compared performance between PRSoS and an existing software (PRSice v1.25) for generating PRS for major depressive disorder using a community cohort (N = 264). We found PRSoS to perform faster than PRSice v1.25 when PRS were generated for a large number of SNPs (~ 17 million SNPs; t = 42.865, p = 5.43E-04). We also show that the use of imputed posterior probabilities and the inclusion of strand-ambiguous SNPs increase the proportion of variance explained by a PRS for major depressive disorder (from 4.3% to 4.8%).
Conclusions: PRSoS provides the user with the ability to generate PRS using an inclusive and efficient approach that considers a larger number of SNPs than conventional approaches. We show that a PRS for major depressive disorder that includes strand-ambiguous SNPs, calculated using PRSoS, accounts for the largest proportion of variance in symptoms of depression in a community cohort, demonstrating the utility of this approach. The availability of this software will help users develop more informative PRS for a variety of complex phenotypes.
Keywords: Bioinformatics; Genetic profile score, Multi-core processing; Major depressive disorder; PRS-on-spark; PRSoS; Polygenic risk score.
Conflict of interest statement
Fully informed written consent was obtained from participants and ethical approval for this study obtained from the Comité d’éthique de la recherche at the Douglas Hospital Research Centre (Montreal, Canada).
Not applicable.
The authors declare that they have no competing interests.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
