

Realizing the significance of noncoding functionality in clinical genomics
- PMID: 30089779
- PMCID: PMC6082831
- DOI: 10.1038/s12276-018-0087-0
Realizing the significance of noncoding functionality in clinical genomics
Abstract
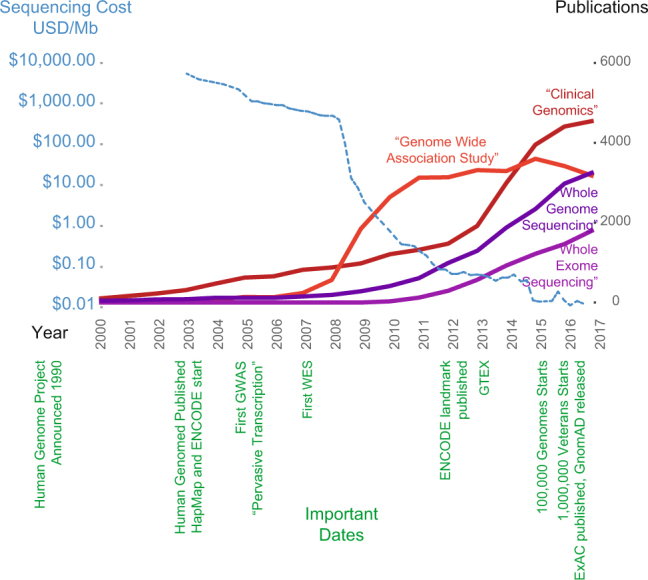
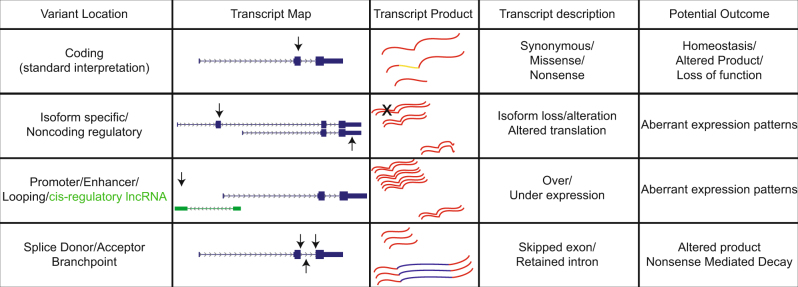
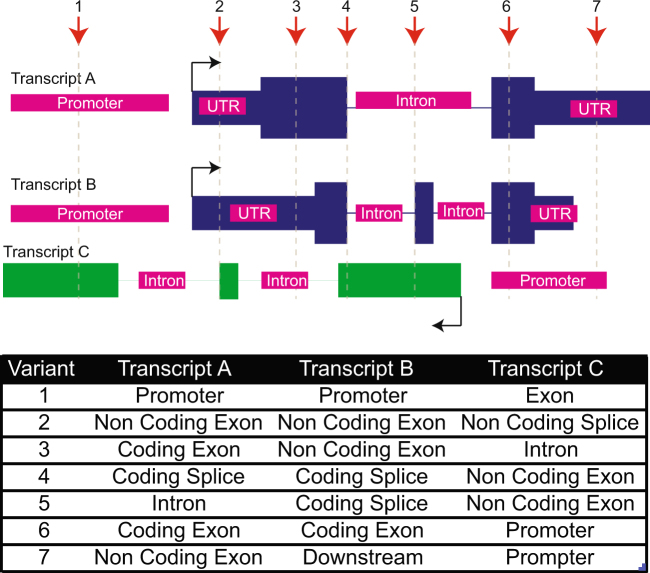
Clinical genomics promises unprecedented precision in understanding the genetic basis of disease. Understanding the impact of variation across the genome is required to realize this potential. Currently, clinical genomics analyses focus on protein-coding genes. However, the noncoding genome is substantially larger than the protein-coding counterpart, and contains structural, regulatory, and transcribed information that needs to be incorporated into genome annotations if the full extent of the opportunity to use genomic information in healthcare is to be realized. This article reviews the challenges and opportunities in unlocking the clinical significance of coding and noncoding genomic information and translating its utility in practice.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
