

Single-cell RNA sequencing technologies and bioinformatics pipelines
- PMID: 30089861
- PMCID: PMC6082860
- DOI: 10.1038/s12276-018-0071-8
Single-cell RNA sequencing technologies and bioinformatics pipelines
Erratum in
-
Author Correction: Single-cell RNA sequencing technologies and bioinformatics pipelines.Exp Mol Med. 2021 May;53(5):1005. doi: 10.1038/s12276-021-00615-w. Exp Mol Med. 2021. PMID: 34045654 Free PMC article. No abstract available.
Abstract
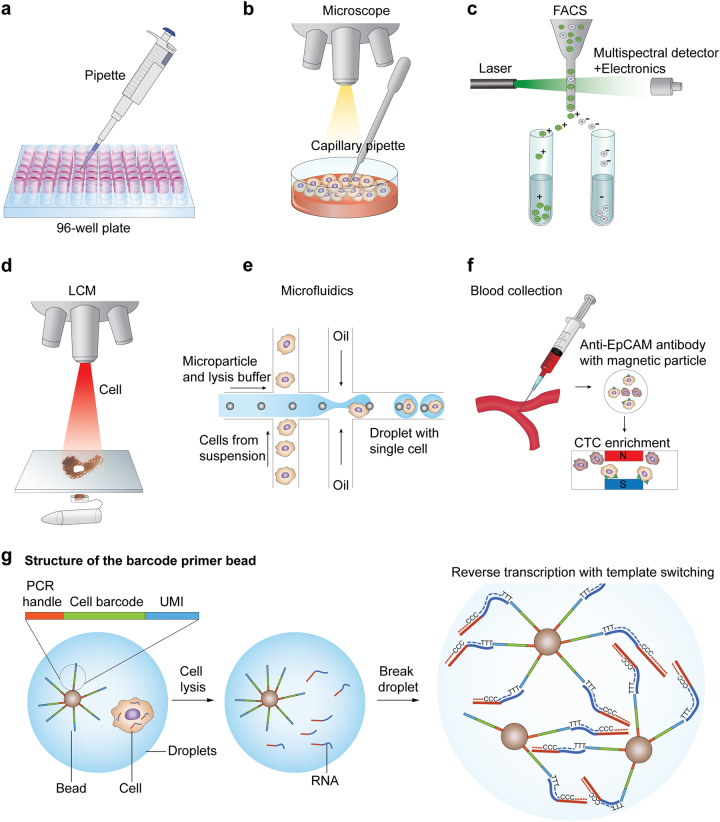
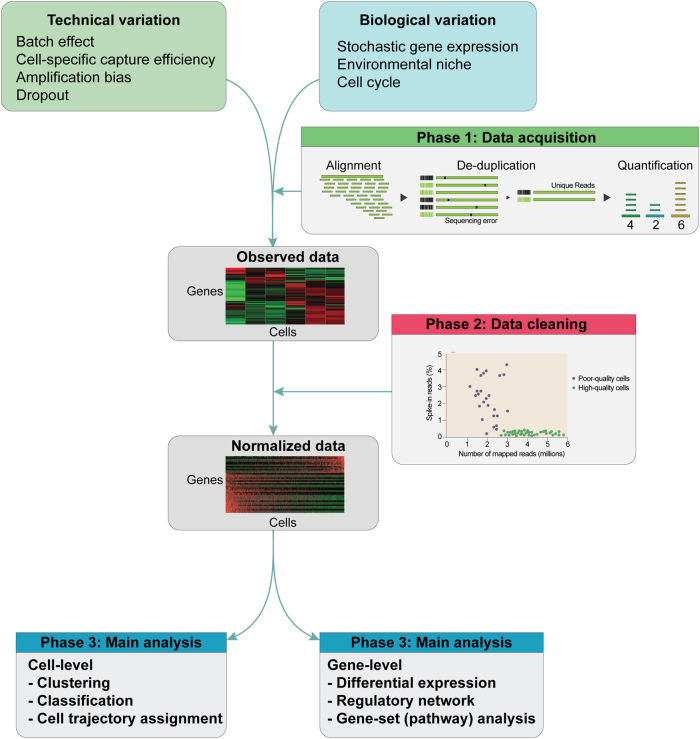
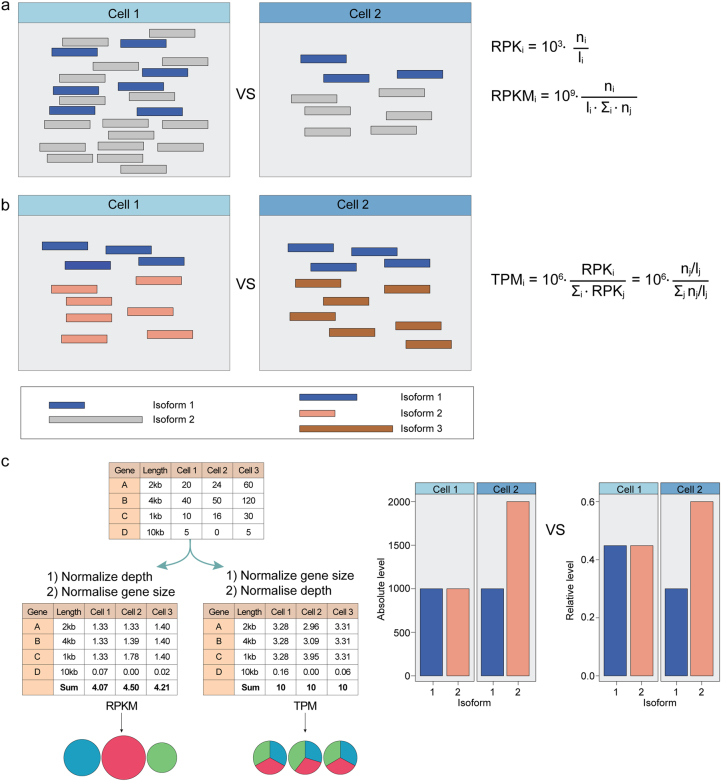
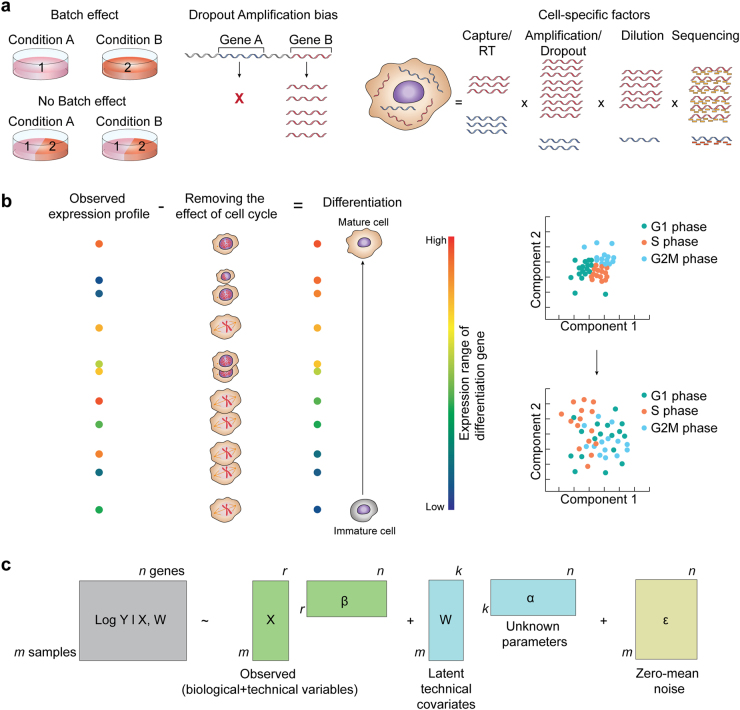
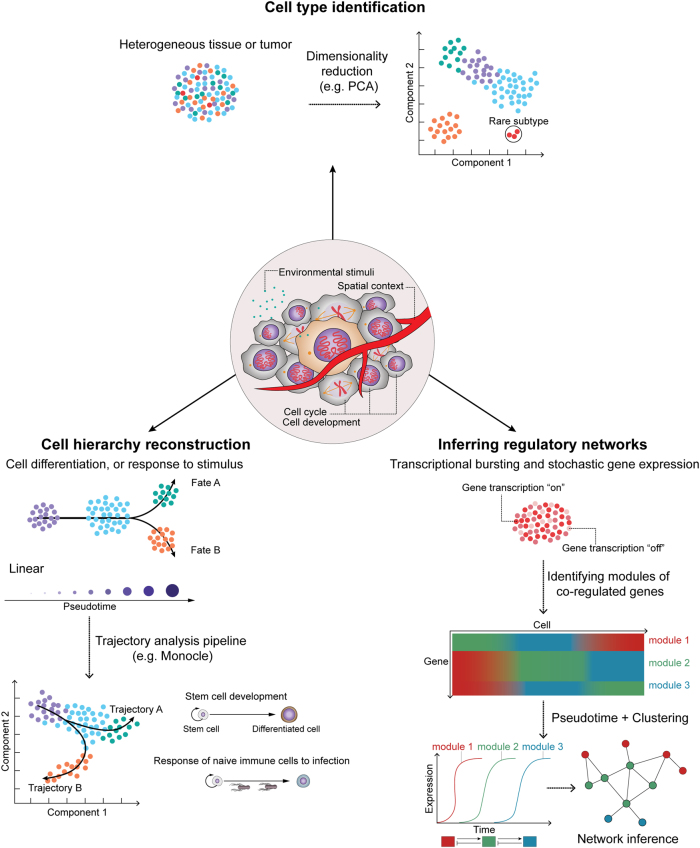
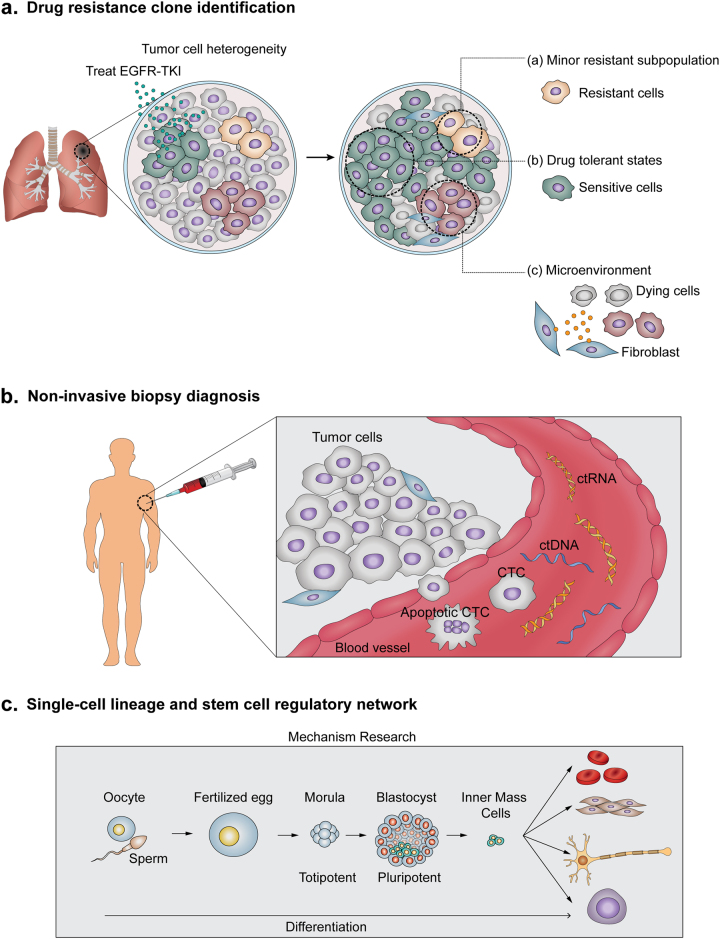
Rapid progress in the development of next-generation sequencing (NGS) technologies in recent years has provided many valuable insights into complex biological systems, ranging from cancer genomics to diverse microbial communities. NGS-based technologies for genomics, transcriptomics, and epigenomics are now increasingly focused on the characterization of individual cells. These single-cell analyses will allow researchers to uncover new and potentially unexpected biological discoveries relative to traditional profiling methods that assess bulk populations. Single-cell RNA sequencing (scRNA-seq), for example, can reveal complex and rare cell populations, uncover regulatory relationships between genes, and track the trajectories of distinct cell lineages in development. In this review, we will focus on technical challenges in single-cell isolation and library preparation and on computational analysis pipelines available for analyzing scRNA-seq data. Further technical improvements at the level of molecular and cell biology and in available bioinformatics tools will greatly facilitate both the basic science and medical applications of these sequencing technologies.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
