

Selective acceleration of disfavored enolate addition reactions by anion-π interactions
- PMID: 30090238
- PMCID: PMC6054047
- DOI: 10.1039/c5sc02563j
Selective acceleration of disfavored enolate addition reactions by anion-π interactions
Abstract
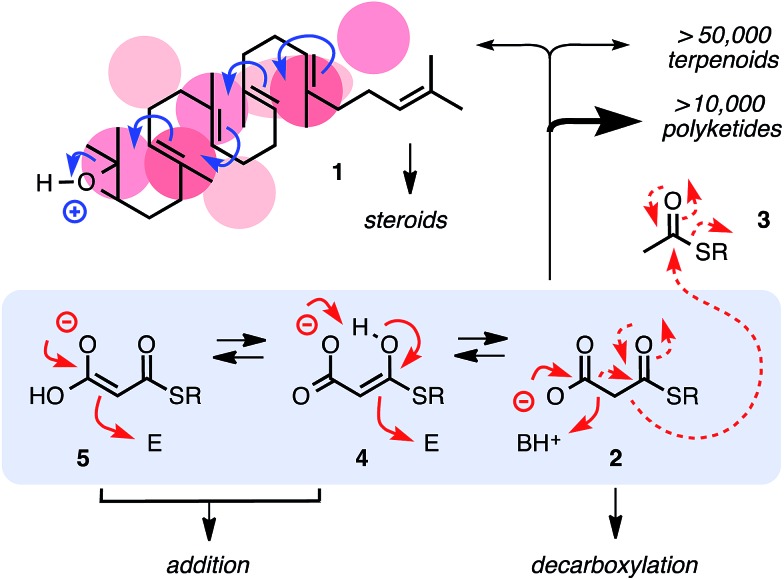
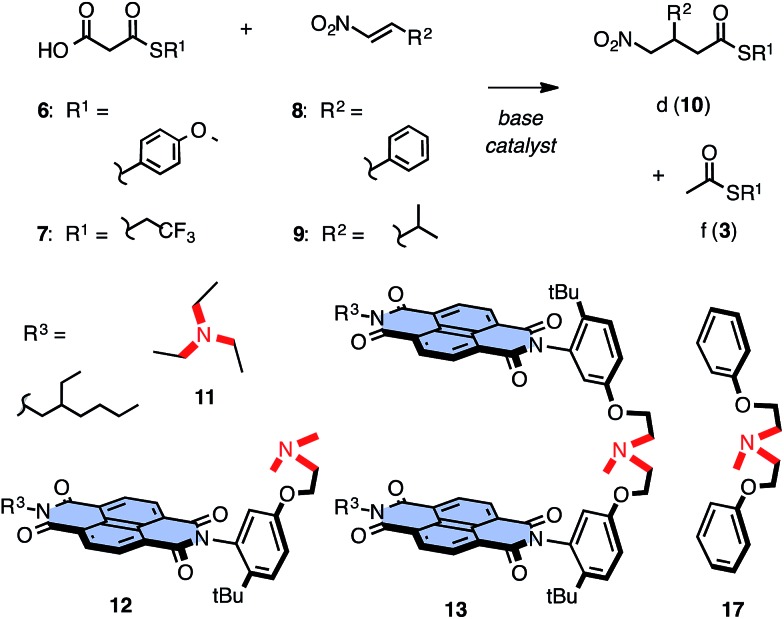
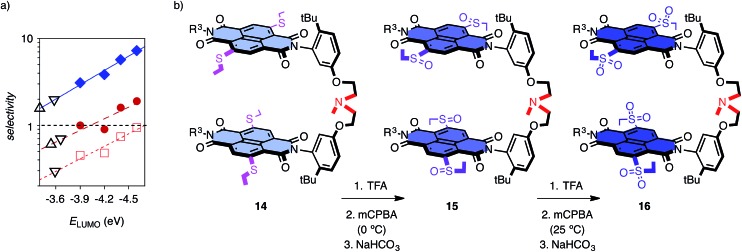
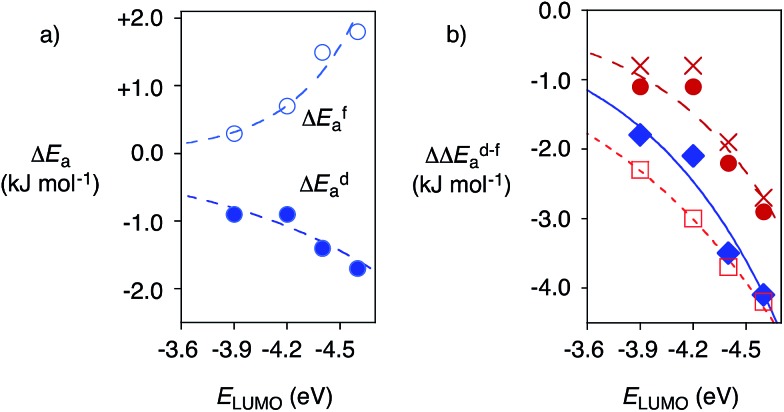
In chemistry and biology, cation-π interactions contribute significantly to many important transformations. In sharp contrast, reactions accomplished with support from the complementary anion-π interactions are essentially unknown. In this report, we show that anion-π interactions can determine the selectivity of the enolate chemistry of malonate half thioesters. Their addition to enolate acceptors is central in natural product biosynthesis but fails without enzymes because non-productive decarboxylation dominates. The newly designed and synthesized anion-π tweezers invert this selectivity by accelerating the disfavored and decelerating the favored process. The discrimination of anionic tautomers of different planarization and charge delocalization on π-acidic surfaces is expected to account for this intriguing "tortoise-and-hare catalysis." Almost exponentially increasing selectivity with increasing π acidity of the catalyst supports that contributions from anion-π interactions are decisive.
Figures
References
-
- Stauffer D. A., Barrans Jr R. E., Dougherty D. A. Angew. Chem., Int. Ed. 1990;29:915–918.
-
- Yamada S., Fossey J. S. Org. Biomol. Chem. 2011;9:7275–7281. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous