

Phylogeny-corrected identification of microbial gene families relevant to human gut colonization
- PMID: 30091981
- PMCID: PMC6084841
- DOI: 10.1371/journal.pcbi.1006242
Phylogeny-corrected identification of microbial gene families relevant to human gut colonization
Abstract
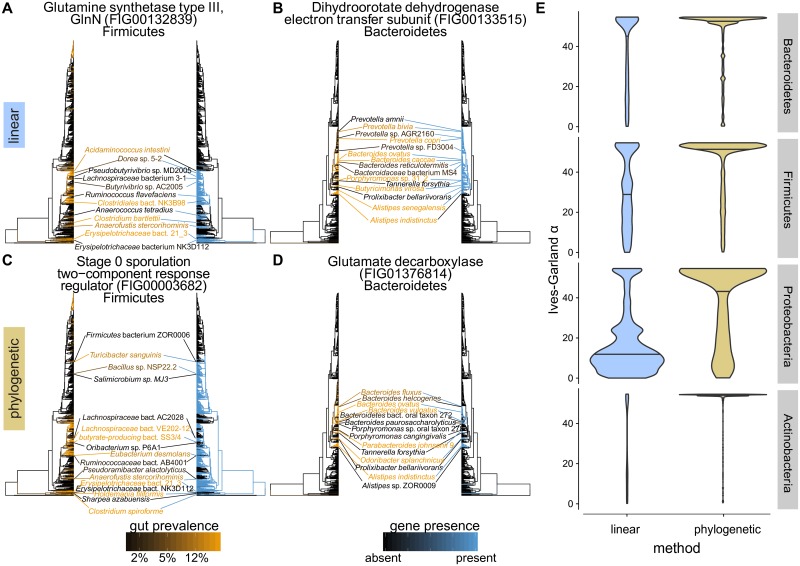
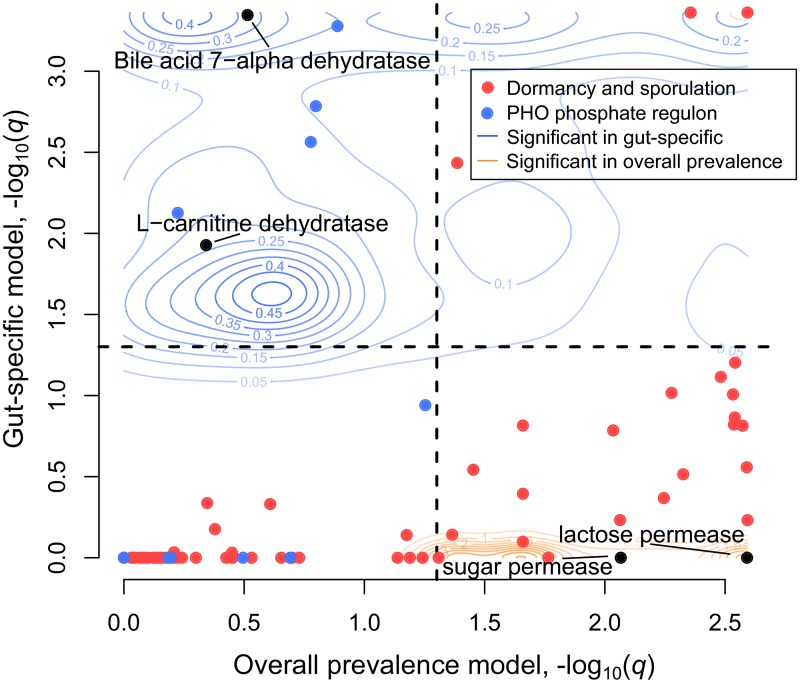
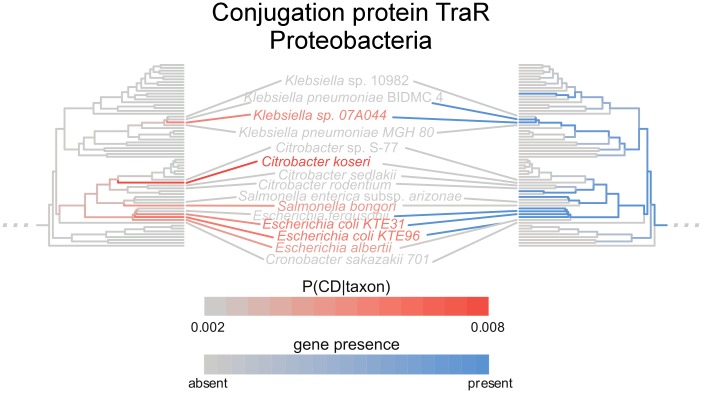
The mechanisms by which different microbes colonize the healthy human gut versus other body sites, the gut in disease states, or other environments remain largely unknown. Identifying microbial genes influencing fitness in the gut could lead to new ways to engineer probiotics or disrupt pathogenesis. We approach this problem by measuring the statistical association between a species having a gene and the probability that the species is present in the gut microbiome. The challenge is that closely related species tend to be jointly present or absent in the microbiome and also share many genes, only a subset of which are involved in gut adaptation. We show that this phylogenetic correlation indeed leads to many false discoveries and propose phylogenetic linear regression as a powerful solution. To apply this method across the bacterial tree of life, where most species have not been experimentally phenotyped, we use metagenomes from hundreds of people to quantify each species' prevalence in and specificity for the gut microbiome. This analysis reveals thousands of genes potentially involved in adaptation to the gut across species, including many novel candidates as well as processes known to contribute to fitness of gut bacteria, such as acid tolerance in Bacteroidetes and sporulation in Firmicutes. We also find microbial genes associated with a preference for the gut over other body sites, which are significantly enriched for genes linked to fitness in an in vivo competition experiment. Finally, we identify gene families associated with higher prevalence in patients with Crohn's disease, including Proteobacterial genes involved in conjugation and fimbria regulation, processes previously linked to inflammation. These gene targets may represent new avenues for modulating host colonization and disease. Our strategy of combining metagenomics with phylogenetic modeling is general and can be used to identify genes associated with adaptation to any environment.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
