

Single-cell RNAseq for the study of isoforms-how is that possible?
- PMID: 30097058
- PMCID: PMC6085759
- DOI: 10.1186/s13059-018-1496-z
Single-cell RNAseq for the study of isoforms-how is that possible?
Abstract
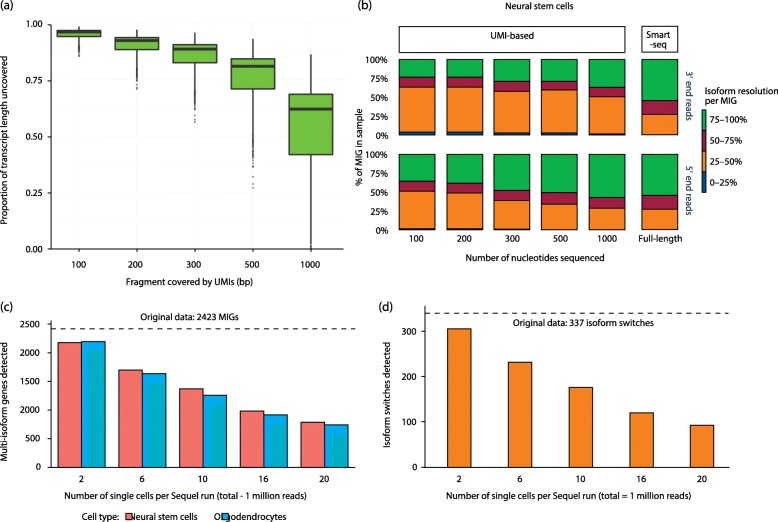
Single-cell RNAseq and alternative splicing studies have recently become two of the most prominent applications of RNAseq. However, the combination of both is still challenging, and few research efforts have been dedicated to the intersection between them. Cell-level insight on isoform expression is required to fully understand the biology of alternative splicing, but it is still an open question to what extent isoform expression analysis at the single-cell level is actually feasible. Here, we establish a set of four conditions that are required for a successful single-cell-level isoform study and evaluate how these conditions are met by these technologies in published research.
Conflict of interest statement
The authors declare that they have no competing interests.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
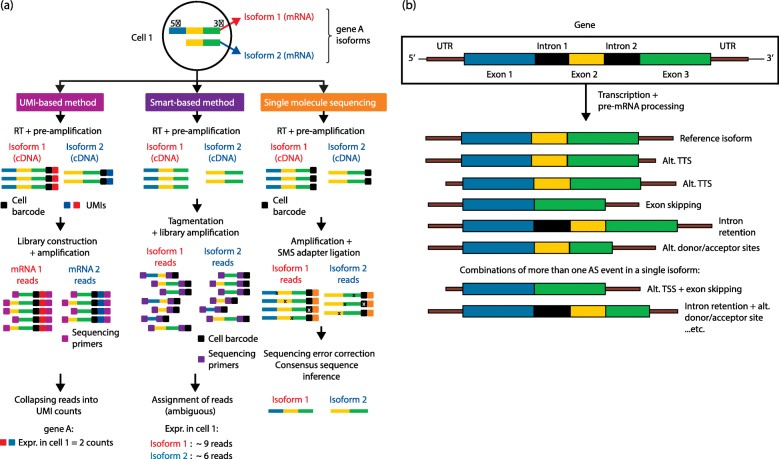
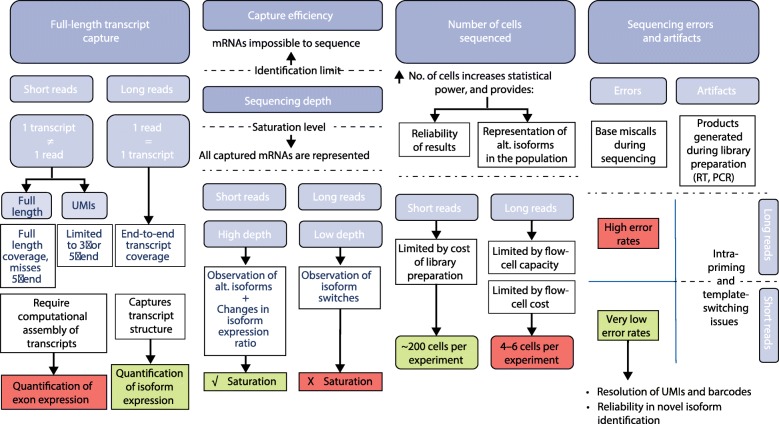
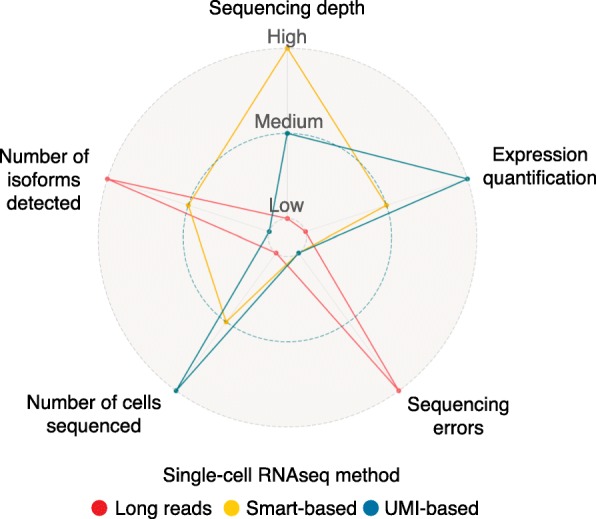
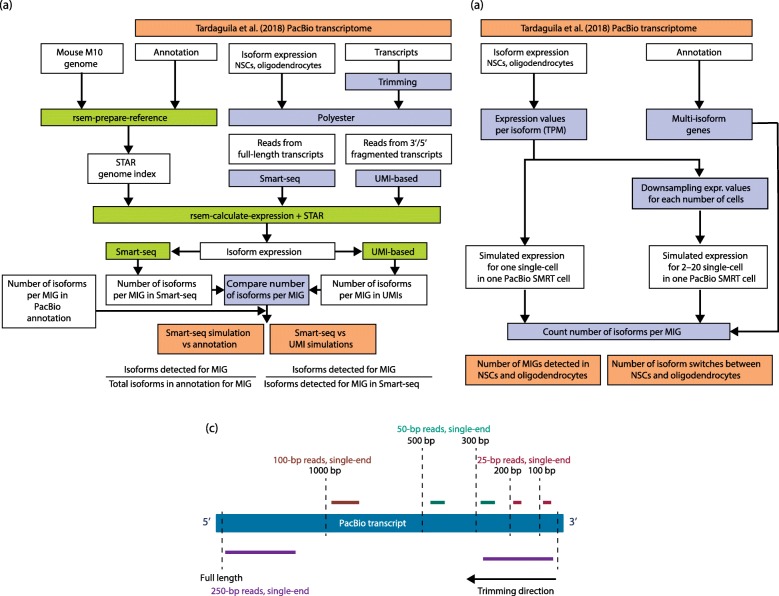
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
