

Maximal viral information recovery from sequence data using VirMAP
- PMID: 30097567
- PMCID: PMC6086868
- DOI: 10.1038/s41467-018-05658-8
Maximal viral information recovery from sequence data using VirMAP
Abstract
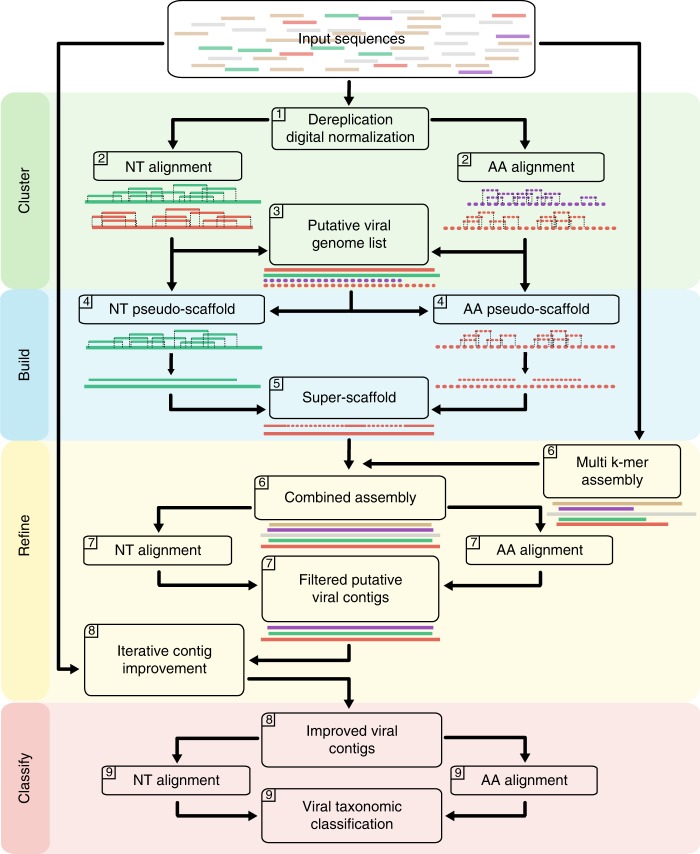
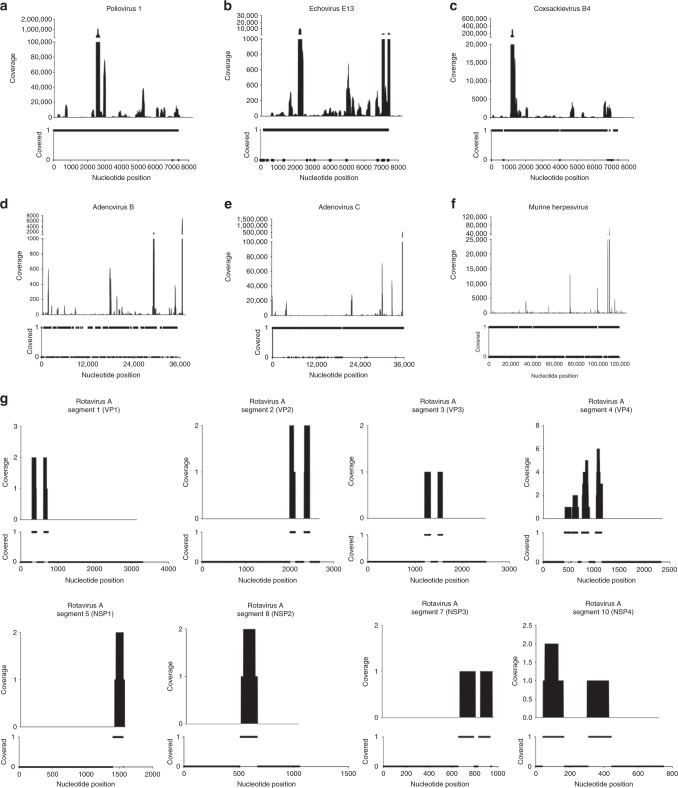
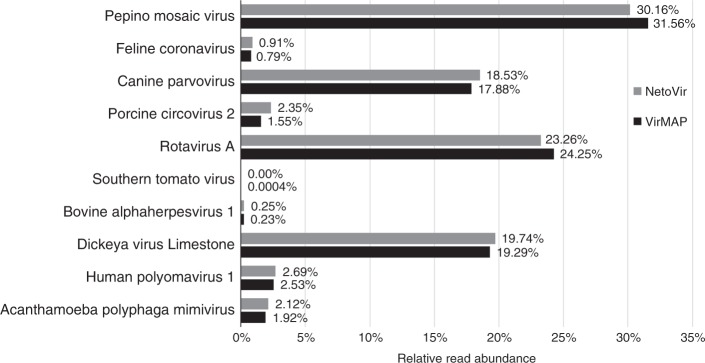
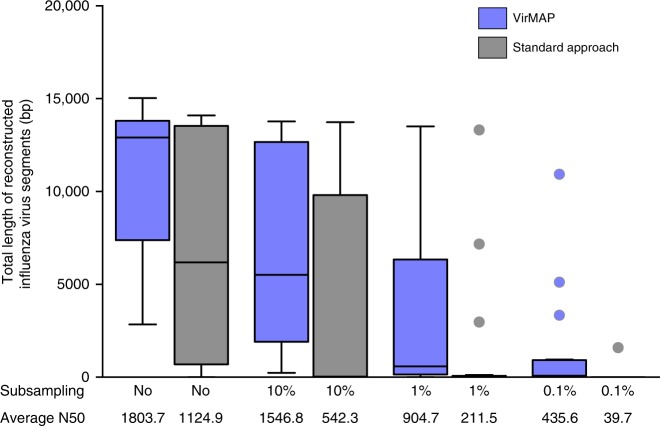
Accurate classification of the human virome is critical to a full understanding of the role viruses play in health and disease. This implies the need for sensitive, specific, and practical pipelines that return precise outputs while still enabling case-specific post hoc analysis. Viral taxonomic characterization from metagenomic data suffers from high background noise and signal crosstalk that confounds current methods. Here we develop VirMAP that overcomes these limitations using techniques that merge nucleotide and protein information to taxonomically classify viral reconstructions independent of genome coverage or read overlap. We validate VirMAP using published data sets and viral mock communities containing RNA and DNA viruses and bacteriophages. VirMAP offers opportunities to enhance metagenomic studies seeking to define virome-host interactions, improve biosurveillance capabilities, and strengthen molecular epidemiology reporting.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
