

Development of an evidence-based algorithm that optimizes sensitivity and specificity in ES-based diagnostics of a clinically heterogeneous patient population
- PMID: 30100613
- PMCID: PMC6752300
- DOI: 10.1038/s41436-018-0016-6
Development of an evidence-based algorithm that optimizes sensitivity and specificity in ES-based diagnostics of a clinically heterogeneous patient population
Abstract
Purpose: Next-generation sequencing (NGS) is rapidly replacing Sanger sequencing in genetic diagnostics. Sensitivity and specificity of NGS approaches are not well-defined, but can be estimated from applying NGS and Sanger sequencing in parallel. Utilizing this strategy, we aimed at optimizing exome sequencing (ES)-based diagnostics of a clinically diverse patient population.
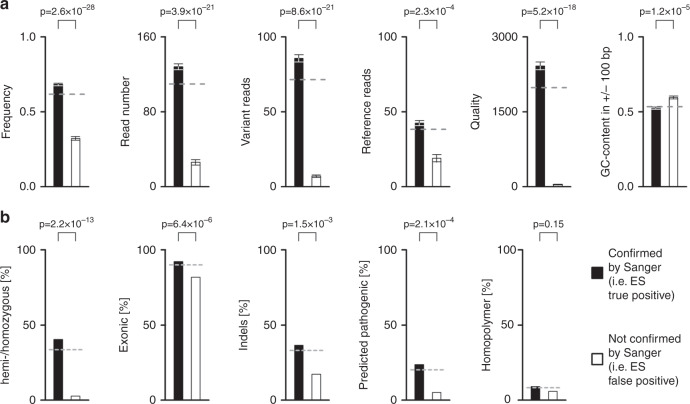
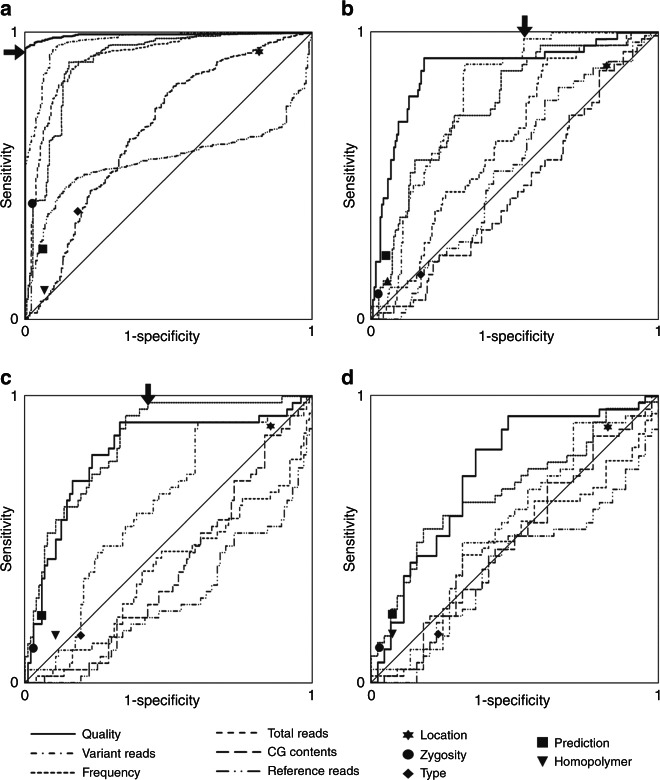
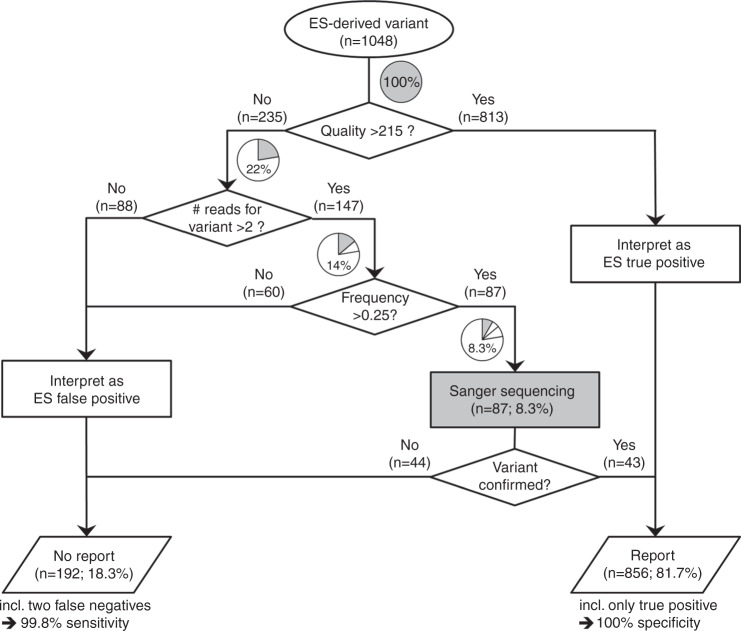
Methods: Consecutive DNA samples from unrelated patients with suspected genetic disease were exome-sequenced; comparatively nonstringent criteria were applied in variant calling. One thousand forty-eight variants in genes compatible with the clinical diagnosis were followed up by Sanger sequencing. Based on a set of variant-specific features, predictors for true positives and true negatives were developed.
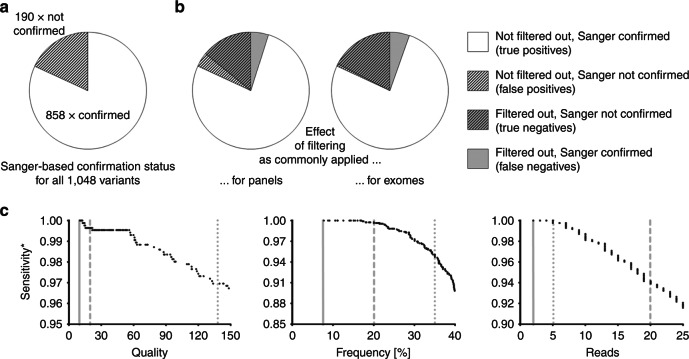
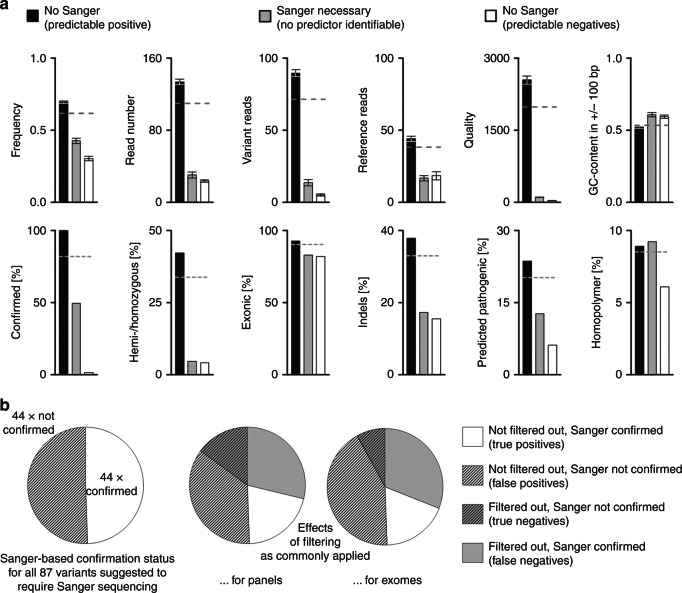
Results: Sanger sequencing confirmed 81.9% of ES-derived variants. Calls from the lower end of stringency accounted for the majority of the false positives, but also contained ~5% of the true positives. A predictor incorporating three variant-specific features classified 91.7% of variants with 100% specificity and 99.75% sensitivity. Confirmation status of the remaining variants (8.3%) was not predictable.
Conclusions: Criteria for variant calling in ES-based diagnostics impact on specificity and sensitivity. Confirmatory sequencing for a proportion of variants, therefore, remains a necessity. Our study exemplifies how these variants can be defined on an empirical basis.
Keywords: Genetic testing; Laboratory standards; Sensitivity; Specificity; exome sequencing.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
