

An approximate Bayesian significance test for genomic evaluations
- PMID: 30101421
- PMCID: PMC6282823
- DOI: 10.1002/bimj.201700219
An approximate Bayesian significance test for genomic evaluations
Abstract
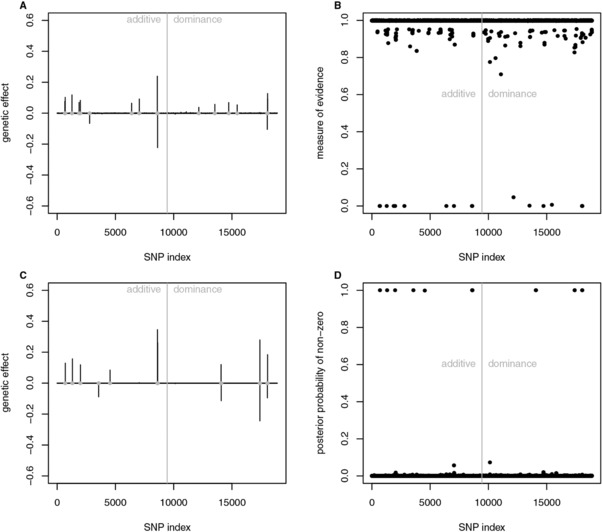
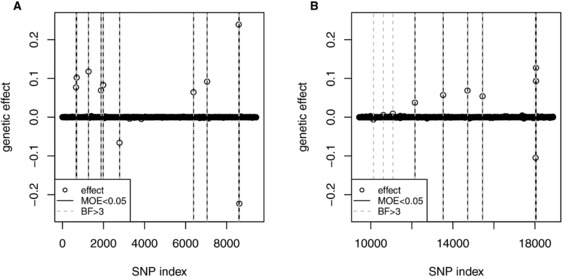
Genomic information can be used to study the genetic architecture of some trait. Not only the size of the genetic effect captured by molecular markers and their position on the genome but also the mode of inheritance, which might be additive or dominant, and the presence of interactions are interesting parameters. When searching for interacting loci, estimating the effect size and determining the significant marker pairs increases the computational burden in terms of speed and memory allocation dramatically. This study revisits a rapid Bayesian approach (fastbayes). As a novel contribution, a measure of evidence is derived to select markers with effect significantly different from zero. It is based on the credibility of the highest posterior density interval next to zero in a marginalized manner. This methodology is applied to simulated data resembling a dairy cattle population in order to verify the sensitivity of testing for a given range of type-I error levels. A real data application complements this study. Sensitivity and specificity of fastbayes were similar to a variational Bayesian method, and a further reduction of computing time could be achieved. More than 50% of the simulated causative variants were identified. The most complex model containing different kinds of genetic effects and their pairwise interactions yielded the best outcome over a range of type-I error levels. The validation study showed that fastbayes is a dual-purpose tool for genomic inferences - it is applicable to predict future outcome of not-yet phenotyped individuals with high precision as well as to estimate and test single-marker effects. Furthermore, it allows the estimation of billions of interaction effects.
Keywords: SNP; conditional expectation; dominance; epistasis; genetic architecture.
© 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures
Similar articles
-
A computationally efficient algorithm for genomic prediction using a Bayesian model.Genet Sel Evol. 2015 Apr 30;47(1):34. doi: 10.1186/s12711-014-0082-4. Genet Sel Evol. 2015. PMID: 25926276 Free PMC article.
-
Comparison of genomic predictions using genomic relationship matrices built with different weighting factors to account for locus-specific variances.J Dairy Sci. 2014 Oct;97(10):6547-59. doi: 10.3168/jds.2014-8210. Epub 2014 Aug 14. J Dairy Sci. 2014. PMID: 25129495
-
A multi-trait Bayesian method for mapping QTL and genomic prediction.Genet Sel Evol. 2018 Mar 24;50(1):10. doi: 10.1186/s12711-018-0377-y. Genet Sel Evol. 2018. PMID: 29571285 Free PMC article.
-
Use of a Bayesian model including QTL markers increases prediction reliability when test animals are distant from the reference population.J Dairy Sci. 2019 Aug;102(8):7237-7247. doi: 10.3168/jds.2018-15815. Epub 2019 May 31. J Dairy Sci. 2019. PMID: 31155255
-
Genome-wide prediction using Bayesian additive regression trees.Genet Sel Evol. 2016 Jun 10;48(1):42. doi: 10.1186/s12711-016-0219-8. Genet Sel Evol. 2016. PMID: 27286957 Free PMC article.
References
-
- Chen, C. , & Tempelman, R. (2015). An integrated approach to empirical Bayesian whole genome prediction modeling. Journal of Agricultural, Biological, and Environmental Statistics, 20(4), 491–511.
-
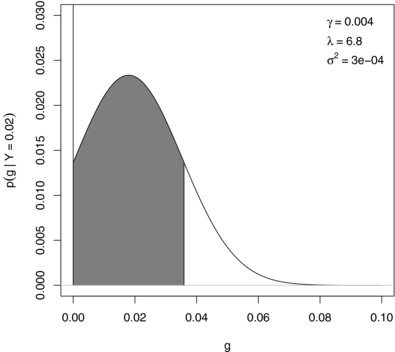
- De Braganca Pereira, C. A. , & Stern, J. M. (1999). Evidence and credibility: Full Bayesian significance test for precise hypotheses. Entropy, 1(4), 99–110.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
