

Bile Microinfarcts in Cholestasis Are Initiated by Rupture of the Apical Hepatocyte Membrane and Cause Shunting of Bile to Sinusoidal Blood
- PMID: 30102412
- PMCID: PMC6587841
- DOI: 10.1002/hep.30213
Bile Microinfarcts in Cholestasis Are Initiated by Rupture of the Apical Hepatocyte Membrane and Cause Shunting of Bile to Sinusoidal Blood
Erratum in
-
Letter to the editor.Arch Toxicol. 2019 Jun;93(6):1771. doi: 10.1007/s00204-019-02472-z. Epub 2019 May 13. Arch Toxicol. 2019. PMID: 31087121 Free PMC article. No abstract available.
Abstract
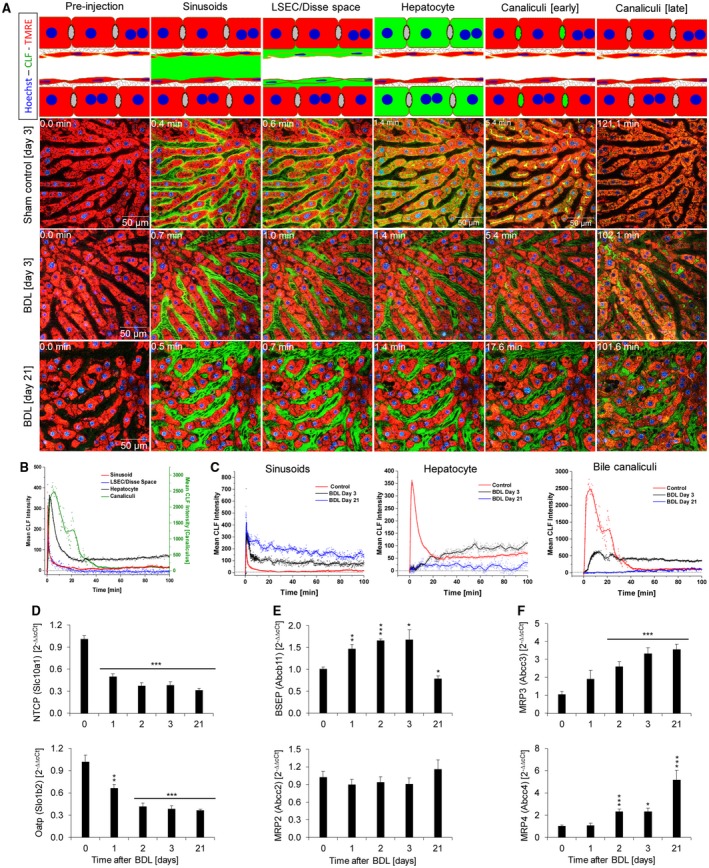
Bile duct ligation (BDL) is an experimental procedure that mimics obstructive cholestatic disease. One of the early consequences of BDL in rodents is the appearance of so-called bile infarcts that correspond to Charcot-Gombault necrosis in human cholestasis. The mechanisms causing bile infarcts and their pathophysiological relevance are unclear. Therefore, intravital two photon-based imaging of BDL mice was performed with fluorescent bile salts (BS) and non-BS organic anion analogues. Key findings were followed up by matrix-assisted laser desorption ionization imaging, clinical chemistry, immunostaining, and gene expression analyses. In the acute phase, 1-3 days after BDL, BS concentrations in bile increased and single-cell bile microinfarcts occurred in dispersed hepatocytes throughout the liver caused by the rupture of the apical hepatocyte membrane. This rupture occurred after loss of mitochondrial membrane potential, followed by entry of bile, cell death, and a "domino effect" of further death events of neighboring hepatocytes. Bile infarcts provided a trans-epithelial shunt between bile canaliculi and sinusoids by which bile constituents leaked into blood. In the chronic phase, ≥21 days after BDL, uptake of BS tracers at the sinusoidal hepatocyte membrane was reduced. This contributes to elevated concentrations of BS in blood and decreased concentrations in the biliary tract. Conclusion: Bile microinfarcts occur in the acute phase after BDL in a limited number of dispersed hepatocytes followed by larger infarcts involving neighboring hepatocytes, and they allow leakage of bile from the BS-overloaded biliary tract into blood, thereby protecting the liver from BS toxicity; in the chronic phase after BDL, reduced sinusoidal BS uptake is a dominant protective factor, and the kidney contributes to the elimination of BS until cholemic nephropathy sets in.
© 2018 The Authors. Hepatology published by Wiley Periodicals, Inc., on behalf of the American Association for the Study of Liver Diseases.
Conflict of interest statement
Dr. Kuepfer is employed by Bayer. Dr. Jansen advises Intercept and NGM. He is on the speakers’ bureau for Falk.
Figures







Comment in
-
Bile Infarcts: New Insights Into the Pathogenesis of Obstructive Cholestasis.Hepatology. 2019 Feb;69(2):473-475. doi: 10.1002/hep.30291. Epub 2019 Jan 16. Hepatology. 2019. PMID: 30251326 No abstract available.
-
Two-photon based imaging reveals mechanisms of tissue damage.Arch Toxicol. 2018 Dec;92(12):3599-3600. doi: 10.1007/s00204-018-2337-2. Epub 2018 Nov 7. Arch Toxicol. 2018. PMID: 30406371 No abstract available.
-
Extrahepatic consequences of cholestasis.Arch Toxicol. 2019 Aug;93(8):2421-2423. doi: 10.1007/s00204-019-02503-9. Epub 2019 Jun 26. Arch Toxicol. 2019. PMID: 31243477 No abstract available.
References
-
- Jansen PL, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, et al. The ascending pathophysiology of cholestatic liver disease. Hepatology 2017;65:722‐738. - PubMed
-
- Harnoss JC, Heidecke CD, Vollmar B, Eipel C. In vivo imaging of bile accumulation and biliary infarction after common bile duct ligation in rats. Eur Surg Res 2011;47:240‐247. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 031A355A/Bundesministerium für Bildung und Forschung/International
- 031L0037/Bundesministerium für Bildung und Forschung/International
- 031L0045/Bundesministerium für Bildung und Forschung/International
- 031L0051/Bundesministerium für Bildung und Forschung/International
- 031L0052/Bundesministerium für Bildung und Forschung/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
