

Role of Resultant Dipole Moment in Mechanical Dissociation of Biological Complexes
- PMID: 30103417
- PMCID: PMC6222447
- DOI: 10.3390/molecules23081995
Role of Resultant Dipole Moment in Mechanical Dissociation of Biological Complexes
Abstract
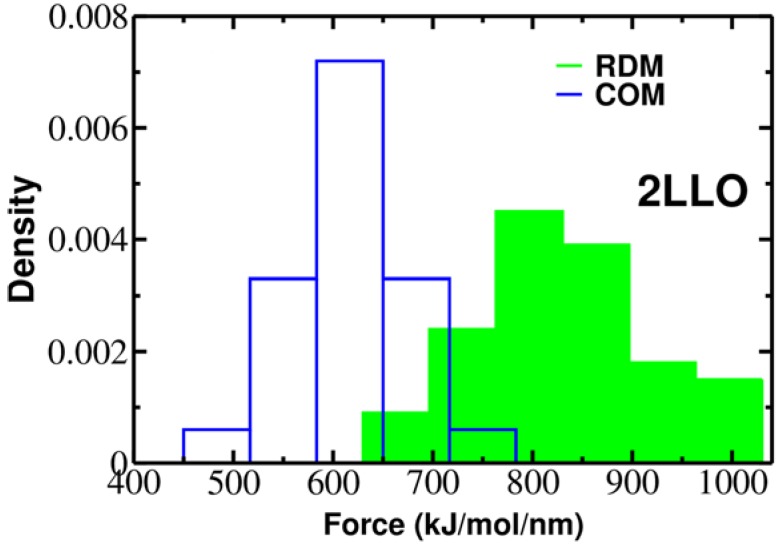
Protein-peptide interactions play essential roles in many cellular processes and their structural characterization is the major focus of current experimental and theoretical research. Two decades ago, it was proposed to employ the steered molecular dynamics (SMD) to assess the strength of protein-peptide interactions. The idea behind using SMD simulations is that the mechanical stability can be used as a promising and an efficient alternative to computationally highly demanding estimation of binding affinity. However, mechanical stability defined as a peak in force-extension profile depends on the choice of the pulling direction. Here we propose an uncommon choice of the pulling direction along resultant dipole moment (RDM) vector, which has not been explored in SMD simulations so far. Using explicit solvent all-atom MD simulations, we apply SMD technique to probe mechanical resistance of ligand-receptor system pulled along two different vectors. A novel pulling direction-when ligand unbinds along the RDM vector-results in stronger forces compared to commonly used ligand unbinding along center of masses vector. Our observation that RDM is one of the factors influencing the mechanical stability of protein-peptide complex can be used to improve the ranking of binding affinities by using mechanical stability as an effective scoring function.
Keywords: all-atom molecular dynamics simulation; mechanical stability; protein-peptide interactions; resultant dipole moment; steered molecular dynamics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Wang L., Wu Y.J., Deng Y.Q., Kim B., Pierce L., Krilov G., Lupyan D., Robinson S., Dahlgren M.K., Greenwood J., et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc. 2015;137:2695–2703. doi: 10.1021/ja512751q. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
