

Deep mutational scanning of hemagglutinin helps predict evolutionary fates of human H3N2 influenza variants
- PMID: 30104379
- PMCID: PMC6126756
- DOI: 10.1073/pnas.1806133115
Deep mutational scanning of hemagglutinin helps predict evolutionary fates of human H3N2 influenza variants
Abstract
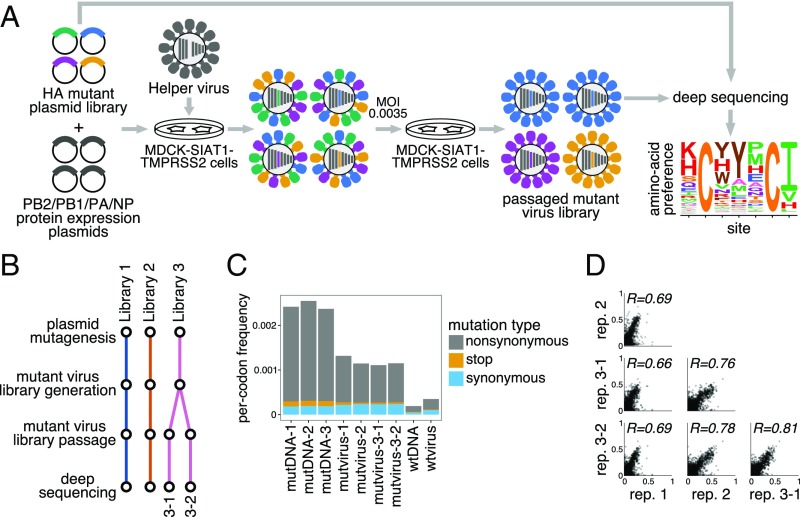
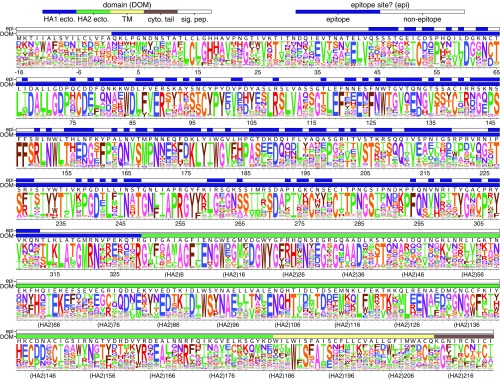
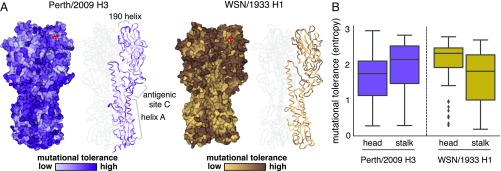
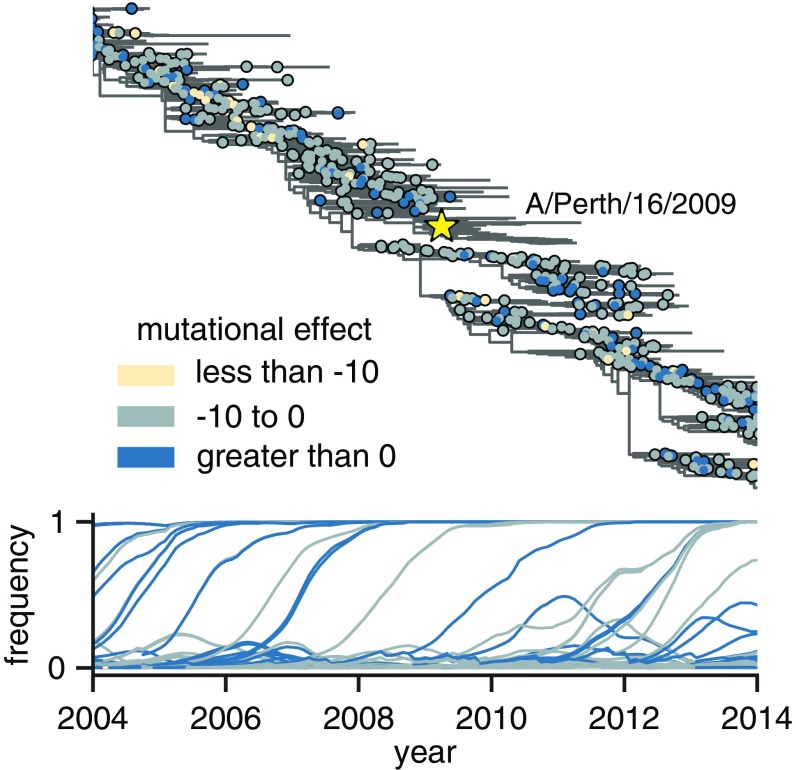
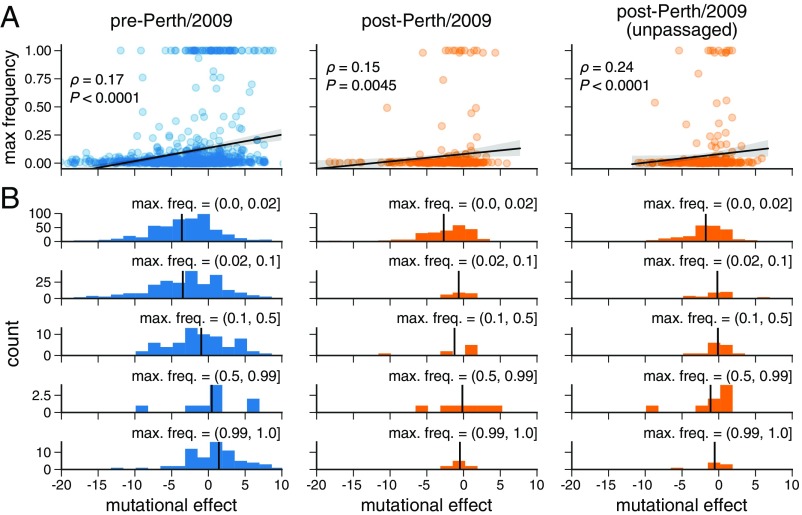
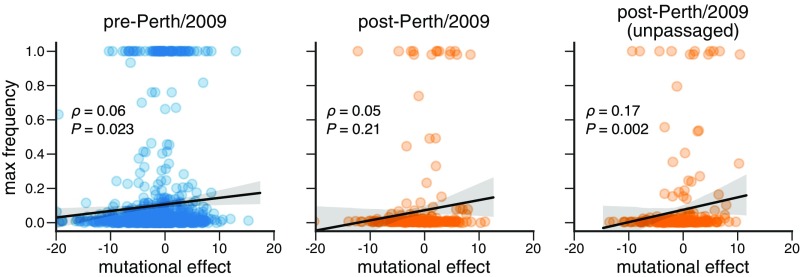
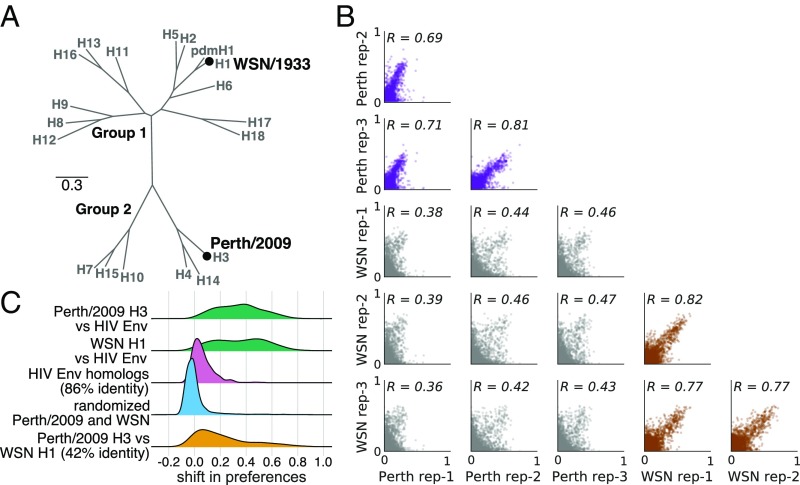
Human influenza virus rapidly accumulates mutations in its major surface protein hemagglutinin (HA). The evolutionary success of influenza virus lineages depends on how these mutations affect HA's functionality and antigenicity. Here we experimentally measure the effects on viral growth in cell culture of all single amino acid mutations to the HA from a recent human H3N2 influenza virus strain. We show that mutations that are measured to be more favorable for viral growth are enriched in evolutionarily successful H3N2 viral lineages relative to mutations that are measured to be less favorable for viral growth. Therefore, despite the well-known caveats about cell-culture measurements of viral fitness, such measurements can still be informative for understanding evolution in nature. We also compare our measurements for H3 HA to similar data previously generated for a distantly related H1 HA and find substantial differences in which amino acids are preferred at many sites. For instance, the H3 HA has less disparity in mutational tolerance between the head and stalk domains than the H1 HA. Overall, our work suggests that experimental measurements of mutational effects can be leveraged to help understand the evolutionary fates of viral lineages in nature-but only when the measurements are made on a viral strain similar to the ones being studied in nature.
Keywords: deep mutational scanning; epistasis; hemagglutinin; influenza virus; mutational shifts.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Smith DJ, et al. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371–376. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
