

Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial
- PMID: 30104418
- PMCID: PMC6088389
- DOI: 10.15252/msb.20178126
Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial
Abstract
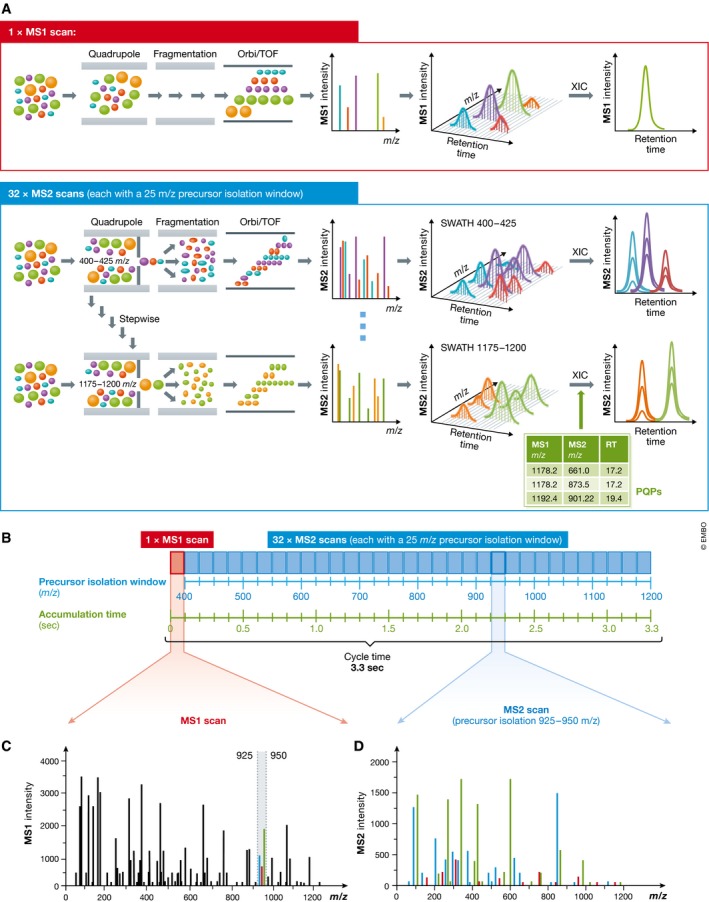
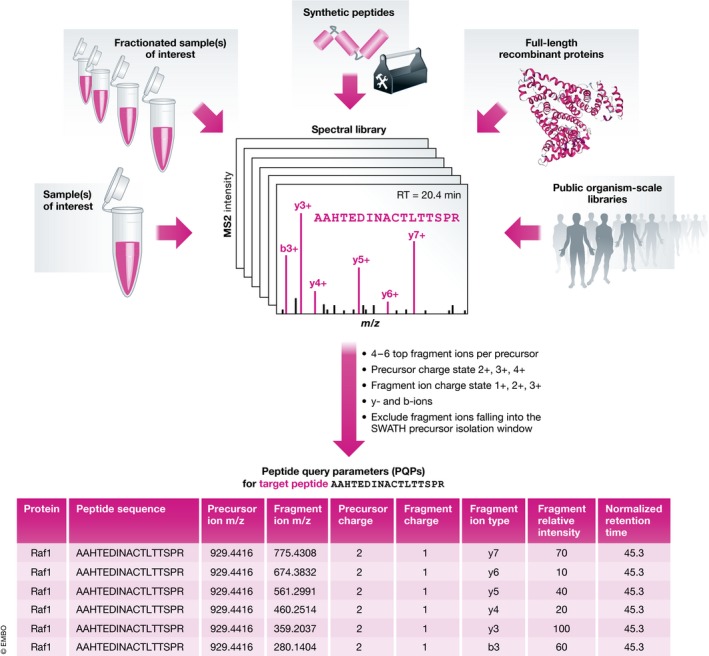
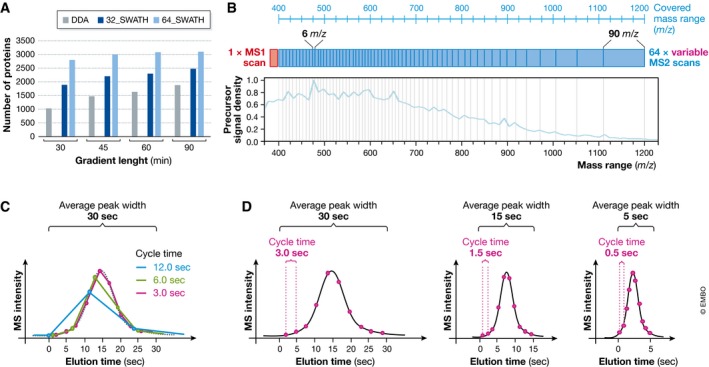
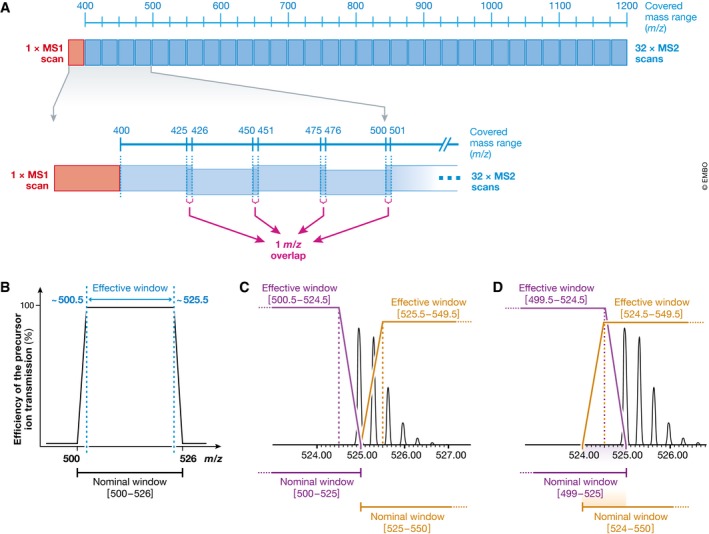
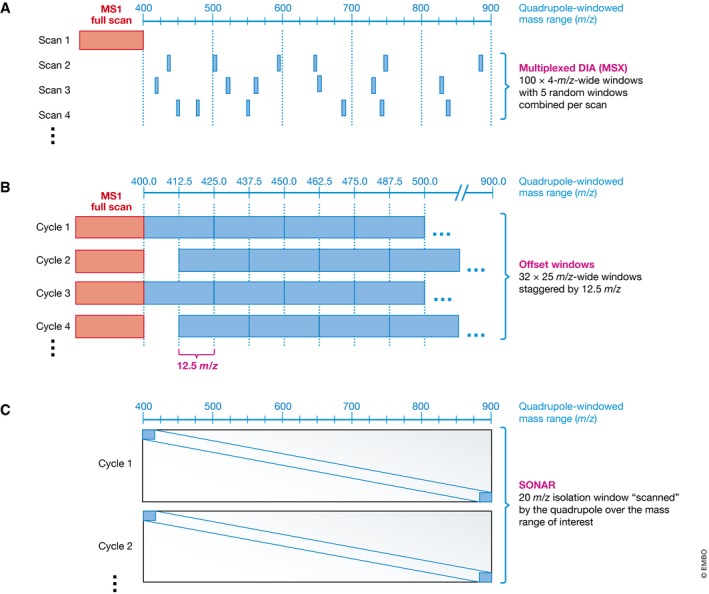
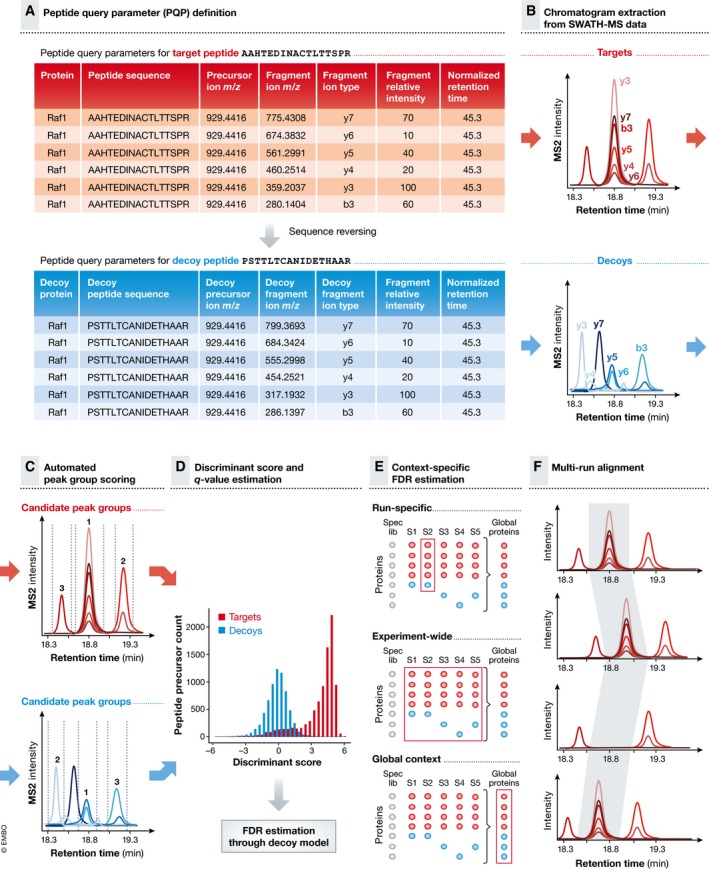
Many research questions in fields such as personalized medicine, drug screens or systems biology depend on obtaining consistent and quantitatively accurate proteomics data from many samples. SWATH-MS is a specific variant of data-independent acquisition (DIA) methods and is emerging as a technology that combines deep proteome coverage capabilities with quantitative consistency and accuracy. In a SWATH-MS measurement, all ionized peptides of a given sample that fall within a specified mass range are fragmented in a systematic and unbiased fashion using rather large precursor isolation windows. To analyse SWATH-MS data, a strategy based on peptide-centric scoring has been established, which typically requires prior knowledge about the chromatographic and mass spectrometric behaviour of peptides of interest in the form of spectral libraries and peptide query parameters. This tutorial provides guidelines on how to set up and plan a SWATH-MS experiment, how to perform the mass spectrometric measurement and how to analyse SWATH-MS data using peptide-centric scoring. Furthermore, concepts on how to improve SWATH-MS data acquisition, potential trade-offs of parameter settings and alternative data analysis strategies are discussed.
Keywords: SWATH‐MS; data‐independent acquisition; mass spectrometry; quantitative proteomics; systems biology.
© 2018 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
References
-
- Aebersold R, Mann M (2016) Mass‐spectrometric exploration of proteome structure and function. Nature 537: 347–355 - PubMed
-
- Ahrne E, Glatter T, Vigano C, Schubert C, Nigg EA, Schmidt A (2016) Evaluation and improvement of quantification accuracy in isobaric mass tag‐based protein quantification experiments. J Proteome Res 15: 2537–2547 - PubMed
-
- Bilbao A, Varesio E, Luban J, Strambio‐De‐Castillia C, Hopfgartner G, Muller M, Lisacek F (2015) Processing strategies and software solutions for data‐independent acquisition in mass spectrometry. Proteomics 15: 964–980 - PubMed
-
- Blainey P, Krzywinski M, Altman N (2014) Points of significance: replication. Nat Methods 11: 879–880 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
