

RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers
- PMID: 30104724
- PMCID: PMC6115280
- DOI: 10.1038/s41556-018-0169-1
RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers
Abstract
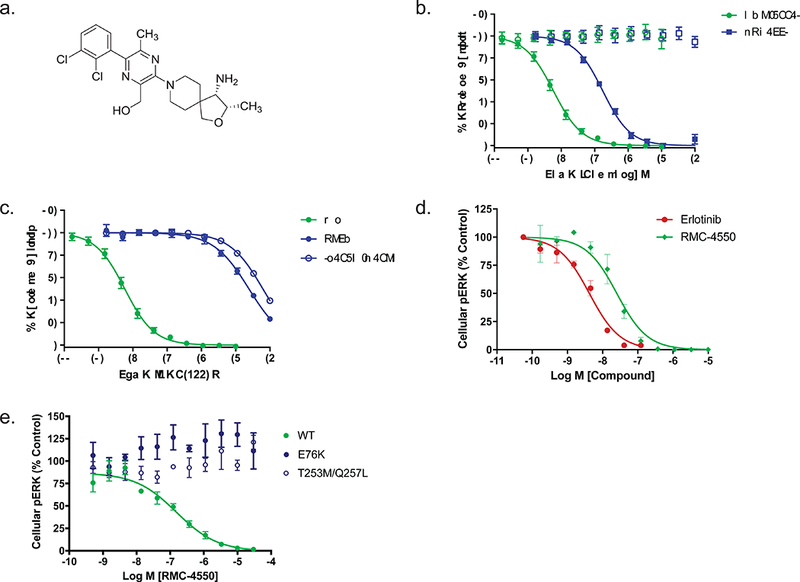
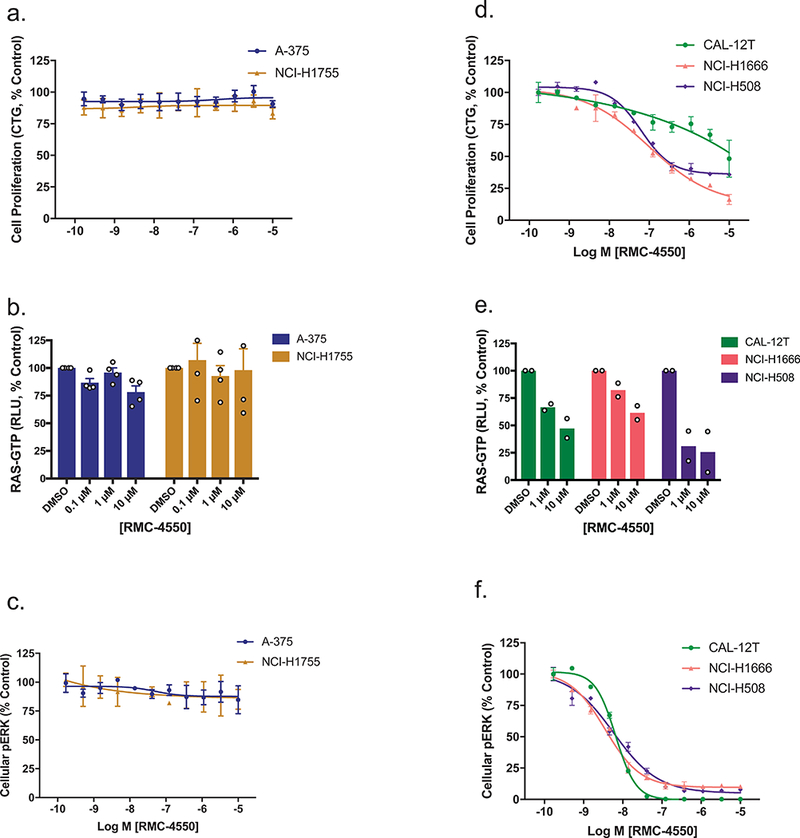
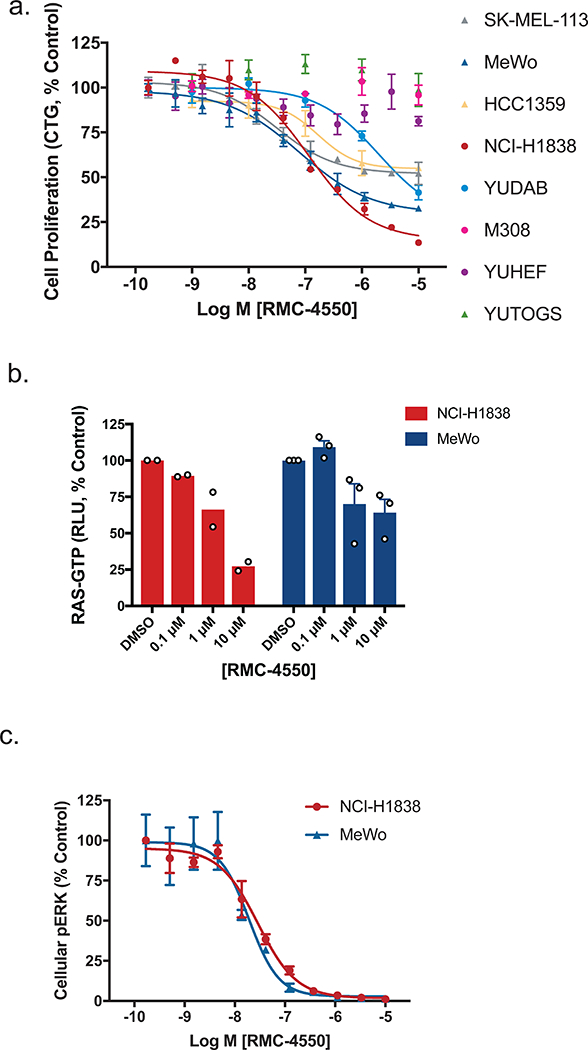
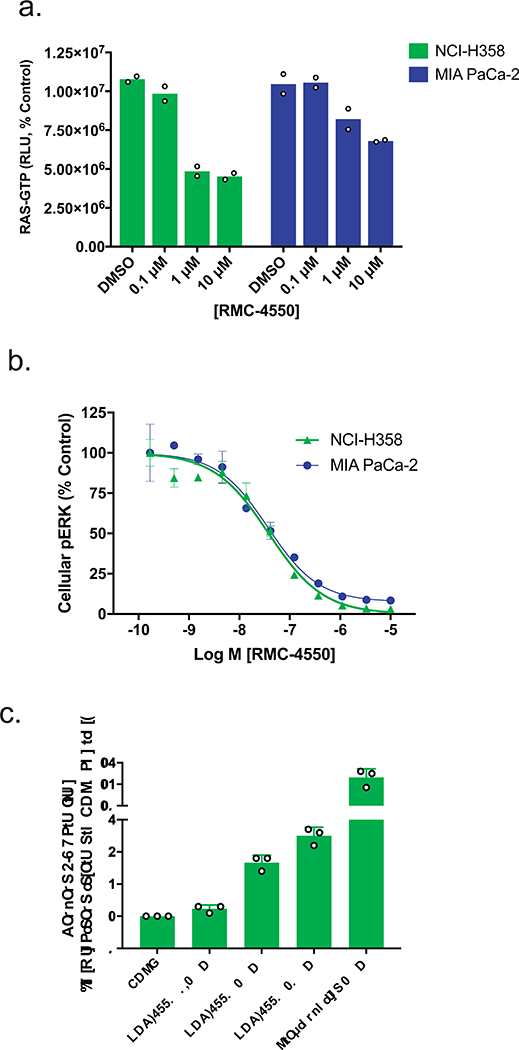
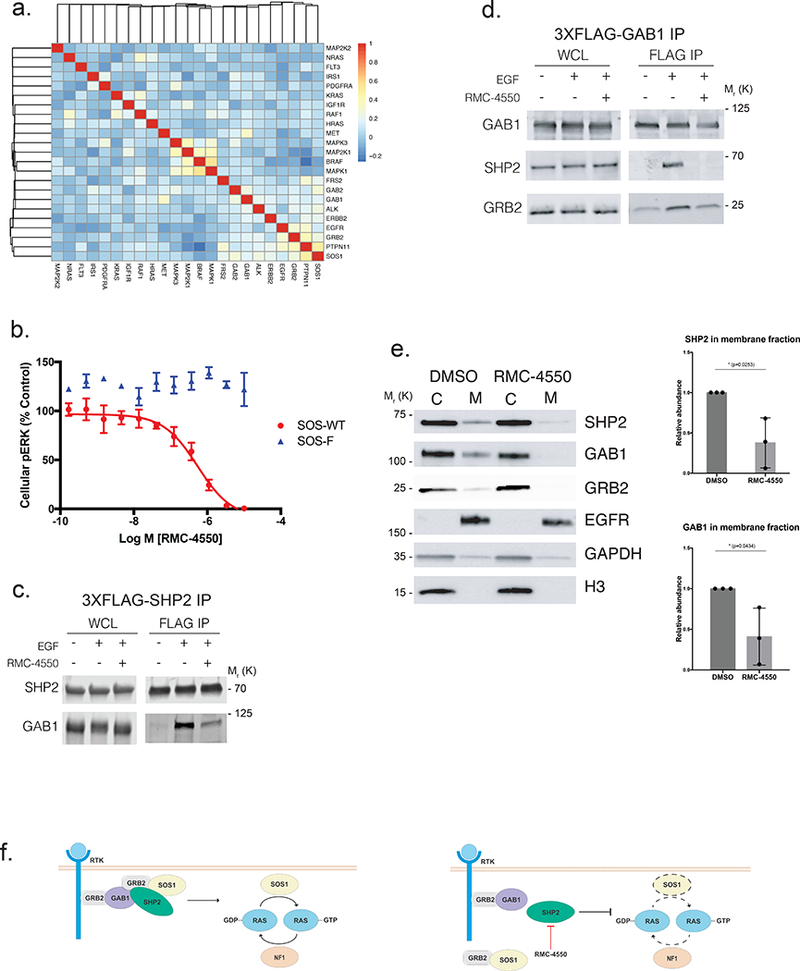
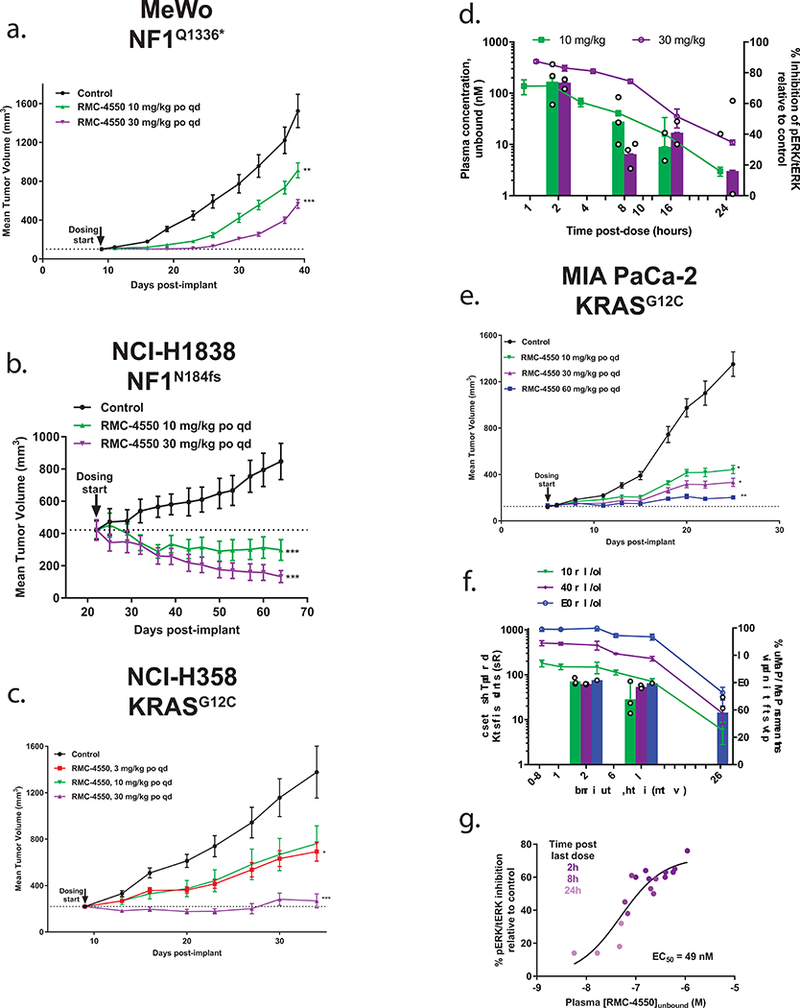
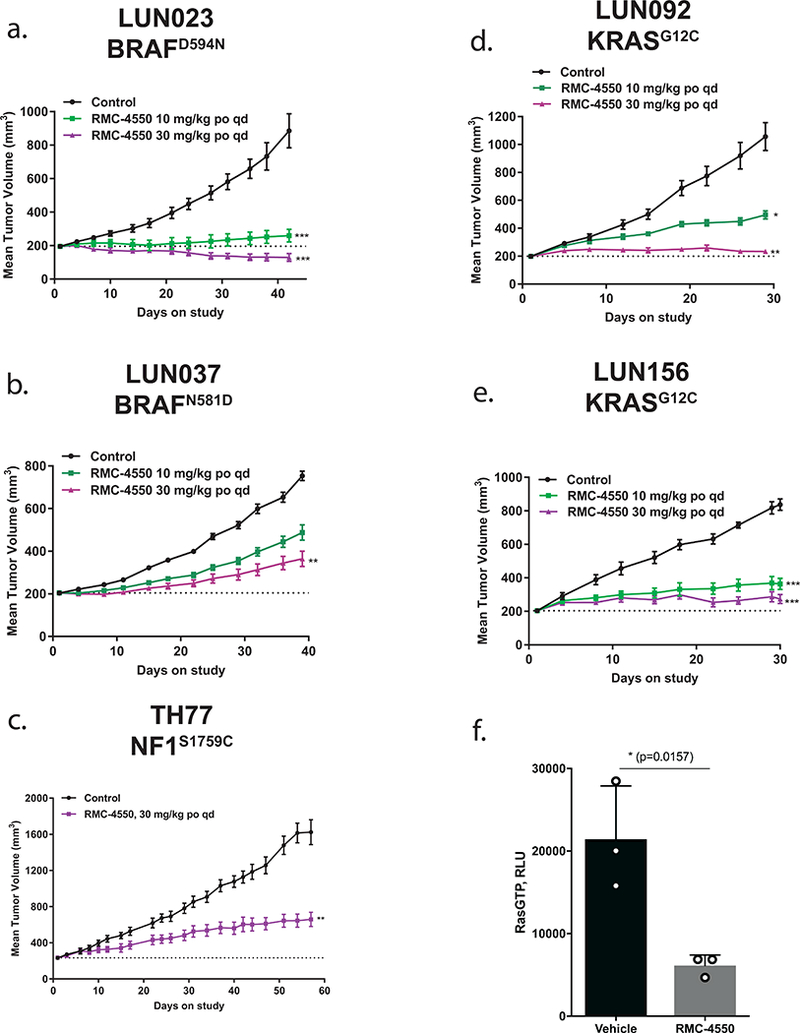
Oncogenic alterations in the RAS/RAF/MEK/ERK pathway drive the growth of a wide spectrum of cancers. While BRAF and MEK inhibitors are efficacious against BRAFV600E-driven cancers, effective targeted therapies are lacking for most cancers driven by other pathway alterations, including non-V600E oncogenic BRAF, RAS GTPase-activating protein (GAP) NF1 (neurofibromin 1) loss and oncogenic KRAS. Here, we show that targeting the SHP2 phosphatase (encoded by PTPN11) with RMC-4550, a small-molecule allosteric inhibitor, is effective in human cancer models bearing RAS-GTP-dependent oncogenic BRAF (for example, class 3 BRAF mutants), NF1 loss or nucleotide-cycling oncogenic RAS (for example, KRASG12C). SHP2 inhibitor treatment decreases oncogenic RAS/RAF/MEK/ERK signalling and cancer growth by disrupting SOS1-mediated RAS-GTP loading. Our findings illuminate a critical function for SHP2 in promoting oncogenic RAS/MAPK pathway activation in cancers with RAS-GTP-dependent oncogenic BRAF, NF1 loss and nucleotide-cycling oncogenic KRAS. SHP2 inhibition is a promising molecular therapeutic strategy for patients with cancers bearing these oncogenic drivers.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
T.G.B. is a consultant to Novartis, AstraZeneca, Array Biopharma (Scientific Advisory Board), Inventiva, and is the recipient of research grants from Ignyta and Revolution Medicines. T.G.B. is a member of the Revolution Medicines Scientific Advisory Board. All Revolution Medicines authors are employees of Revolution Medicines, Inc. The other authors declare no competing financial interests.
Figures
Similar articles
-
PCC0208023, a potent SHP2 allosteric inhibitor, imparts an antitumor effect against KRAS mutant colorectal cancer.Toxicol Appl Pharmacol. 2020 Jul 1;398:115019. doi: 10.1016/j.taap.2020.115019. Epub 2020 Apr 24. Toxicol Appl Pharmacol. 2020. PMID: 32335126
-
RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth.Nature. 2010 Mar 18;464(7287):431-5. doi: 10.1038/nature08833. Epub 2010 Feb 3. Nature. 2010. PMID: 20130576
-
Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling.Clin Cancer Res. 2021 Jan 1;27(1):342-354. doi: 10.1158/1078-0432.CCR-20-2718. Epub 2020 Oct 12. Clin Cancer Res. 2021. PMID: 33046519
-
Targeting oncogenic Raf protein-serine/threonine kinases in human cancers.Pharmacol Res. 2018 Sep;135:239-258. doi: 10.1016/j.phrs.2018.08.013. Epub 2018 Aug 15. Pharmacol Res. 2018. PMID: 30118796 Review.
-
Efficacy of Dabrafenib and Trametinib in a Patient with Squamous-Cell Carcinoma, with Mutation p.D594G in BRAF and p.R461* in NF1 Genes-A Case Report with Literature Review.Int J Mol Sci. 2023 Jan 7;24(2):1195. doi: 10.3390/ijms24021195. Int J Mol Sci. 2023. PMID: 36674722 Free PMC article. Review.
Cited by
-
Extensive preclinical validation of combined RMC-4550 and LY3214996 supports clinical investigation for KRAS mutant pancreatic cancer.Cell Rep Med. 2022 Nov 15;3(11):100815. doi: 10.1016/j.xcrm.2022.100815. Cell Rep Med. 2022. PMID: 36384095 Free PMC article.
-
Overcoming Resistance to Drugs Targeting KRASG12C Mutation.Innovation (Camb). 2020 Aug 28;1(2):100035. doi: 10.1016/j.xinn.2020.100035. Epub 2020 Aug 5. Innovation (Camb). 2020. PMID: 32939510 Free PMC article.
-
Fibroblast growth factor receptor fusions in cancer: opportunities and challenges.J Exp Clin Cancer Res. 2021 Nov 3;40(1):345. doi: 10.1186/s13046-021-02156-6. J Exp Clin Cancer Res. 2021. PMID: 34732230 Free PMC article. Review.
-
Toward a More Precise Future for Oncology.Cancer Cell. 2020 Apr 13;37(4):431-442. doi: 10.1016/j.ccell.2020.03.014. Cancer Cell. 2020. PMID: 32289268 Free PMC article.
-
SHP2 as a Potential Therapeutic Target in Diffuse-Type Gastric Carcinoma Addicted to Receptor Tyrosine Kinase Signaling.Cancers (Basel). 2021 Aug 26;13(17):4309. doi: 10.3390/cancers13174309. Cancers (Basel). 2021. PMID: 34503119 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
