

ASAP - A Webserver for Immunoglobulin-Sequencing Analysis Pipeline
- PMID: 30105017
- PMCID: PMC6077260
- DOI: 10.3389/fimmu.2018.01686
ASAP - A Webserver for Immunoglobulin-Sequencing Analysis Pipeline
Abstract
Reproducible and robust data on antibody repertoires are invaluable for basic and applied immunology. Next-generation sequencing (NGS) of antibody variable regions has emerged as a powerful tool in systems immunology, providing quantitative molecular information on antibody polyclonal composition. However, major computational challenges exist when analyzing antibody sequences, from error handling to hypermutation profiles and clonal expansion analyses. In this work, we developed the ASAP (A webserver for Immunoglobulin-Seq Analysis Pipeline) webserver (https://asap.tau.ac.il). The input to ASAP is a paired-end sequence dataset from one or more replicates, with or without unique molecular identifiers. These datasets can be derived from NGS of human or murine antibody variable regions. ASAP first filters and annotates the sequence reads using public or user-provided germline sequence information. The ASAP webserver next performs various calculations, including somatic hypermutation level, CDR3 lengths, V(D)J family assignments, and V(D)J combination distribution. These analyses are repeated for each replicate. ASAP provides additional information by analyzing the commonalities and differences between the repeats ("joint" analysis). For example, ASAP examines the shared variable regions and their frequency in each replicate to determine which sequences are less likely to be a result of a sample preparation derived and/or sequencing errors. Moreover, ASAP clusters the data to clones and reports the identity and prevalence of top ranking clones (clonal expansion analysis). ASAP further provides the distribution of synonymous and non-synonymous mutations within the V genes somatic hypermutations. Finally, ASAP provides means to process the data for proteomic analysis of serum/secreted antibodies by generating a variable region database for liquid chromatography high resolution tandem mass spectrometry (LC-MS/MS) interpretation. ASAP is user-friendly, free, and open to all users, with no login requirement. ASAP is applicable for researchers interested in basic questions related to B cell development and differentiation, as well as applied researchers who are interested in vaccine development and monoclonal antibody engineering. By virtue of its user-friendliness, ASAP opens the antibody analysis field to non-expert users who seek to boost their research with immune repertoire analysis.
Keywords: AIRR-Seq; B cell receptor; Ig-Seq; antibodies; antibody repertoire analysis; high throughput sequencing; immune repertoire; next generation sequencing.
Figures
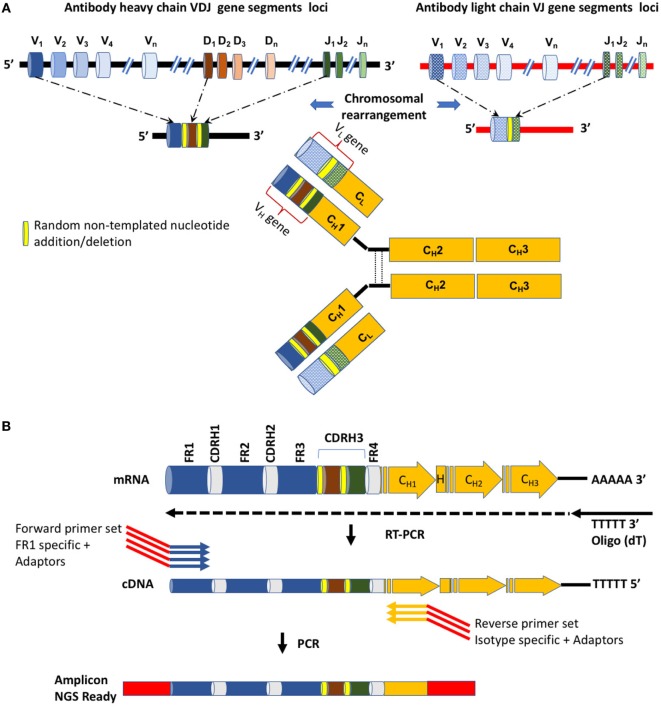

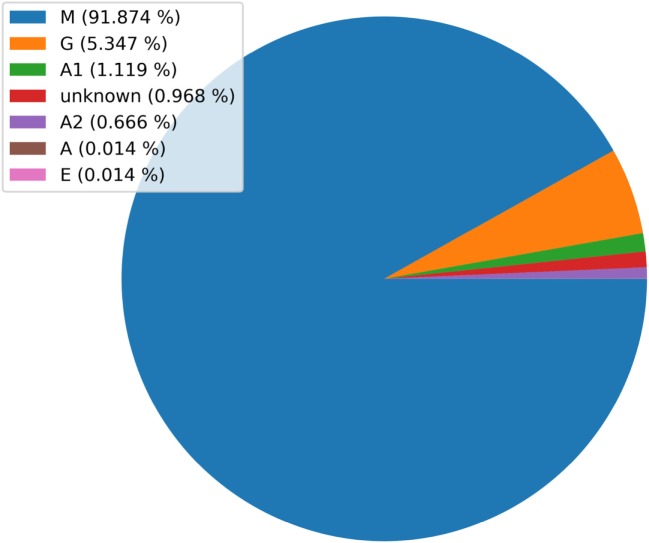
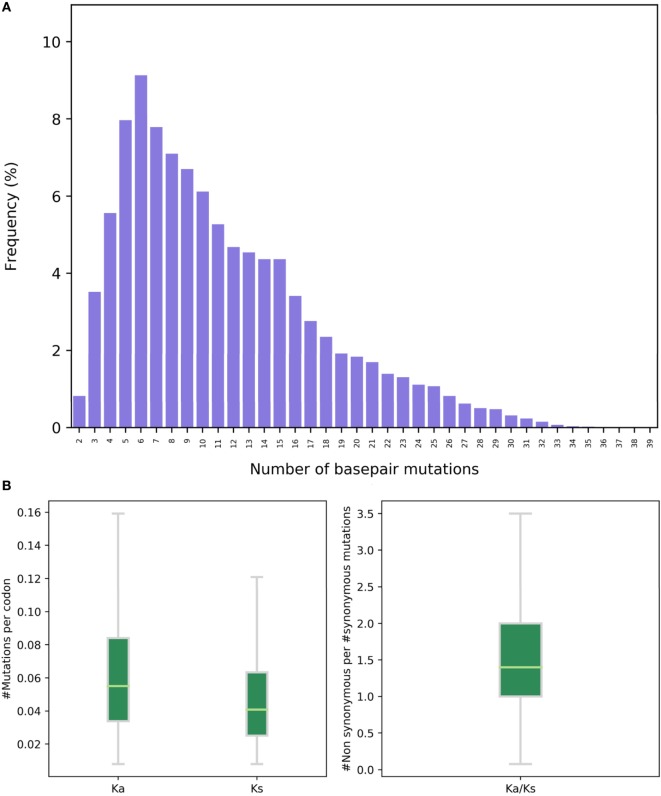
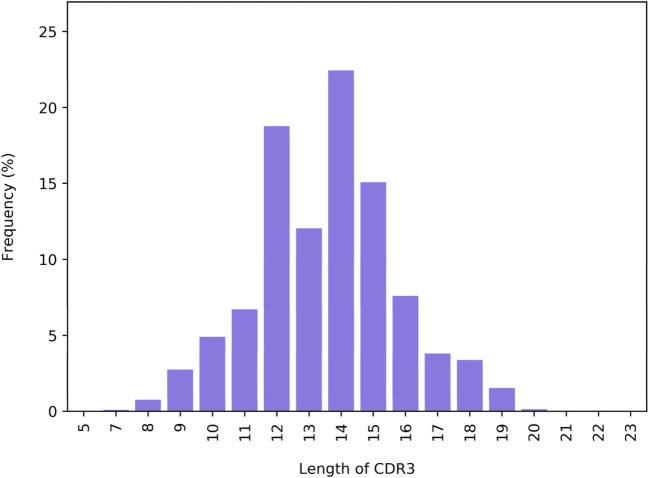
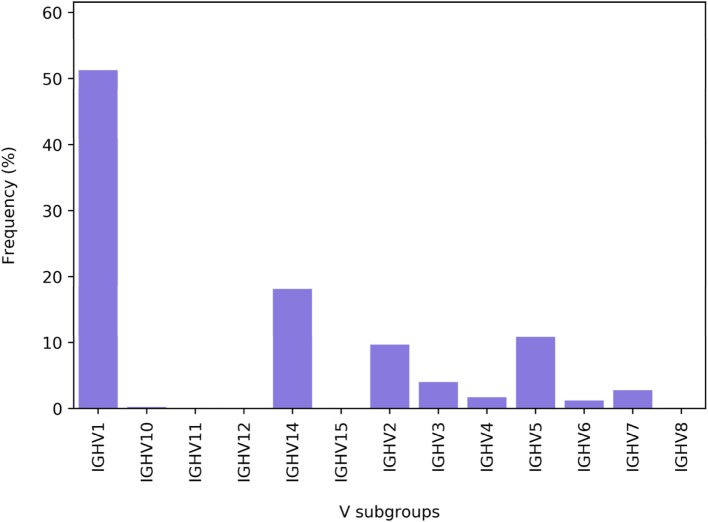
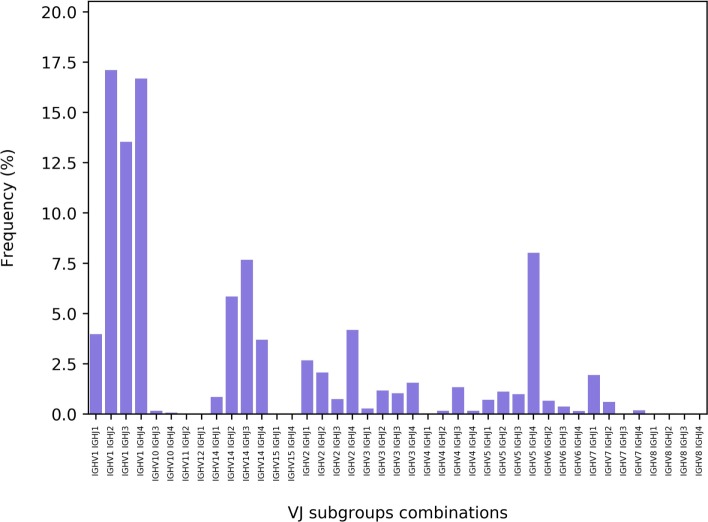
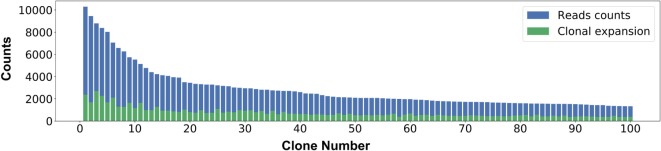

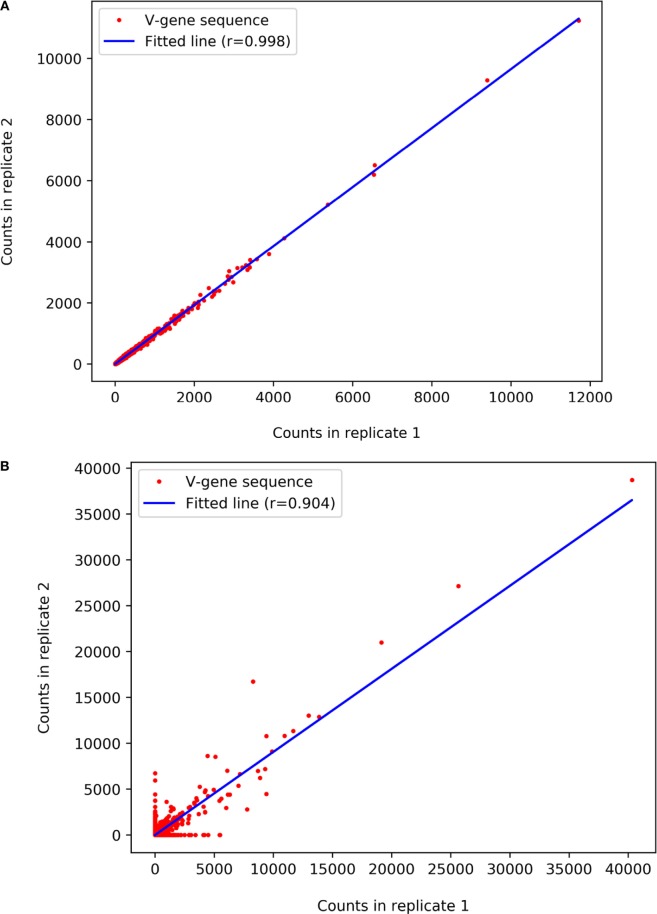
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources