

UBXN2A enhances CHIP-mediated proteasomal degradation of oncoprotein mortalin-2 in cancer cells
- PMID: 30107089
- PMCID: PMC6166003
- DOI: 10.1002/1878-0261.12372
UBXN2A enhances CHIP-mediated proteasomal degradation of oncoprotein mortalin-2 in cancer cells
Abstract
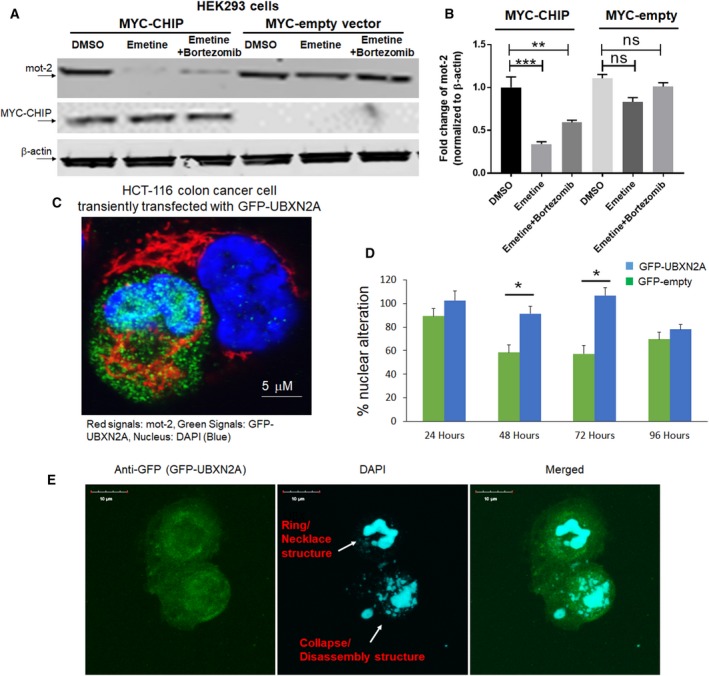
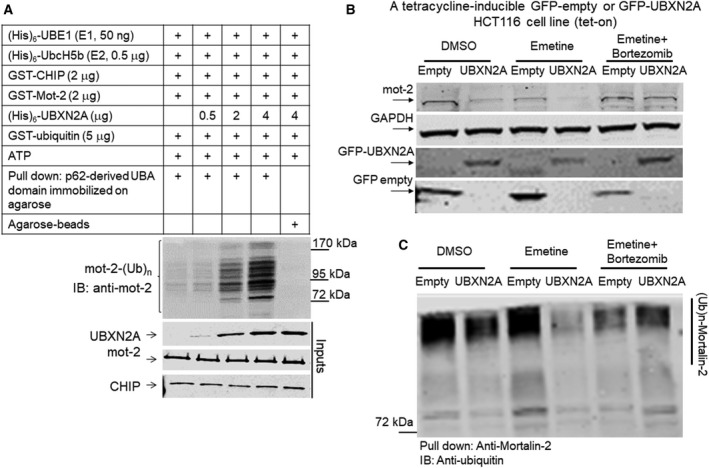
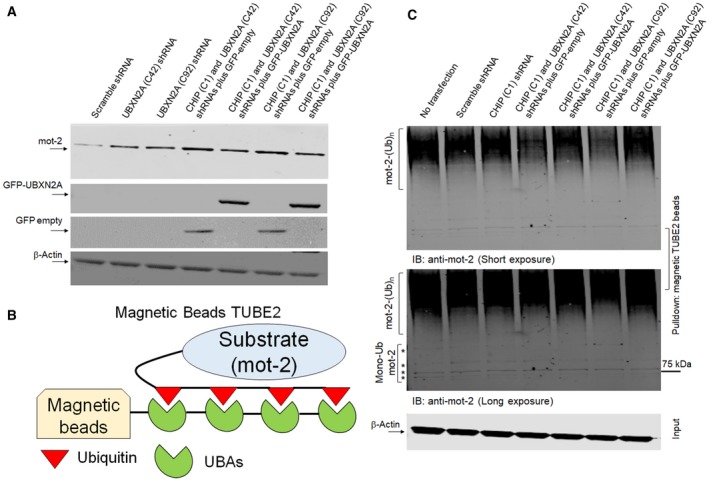
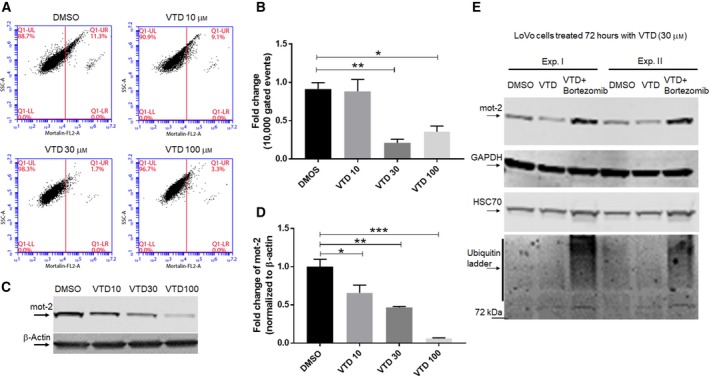
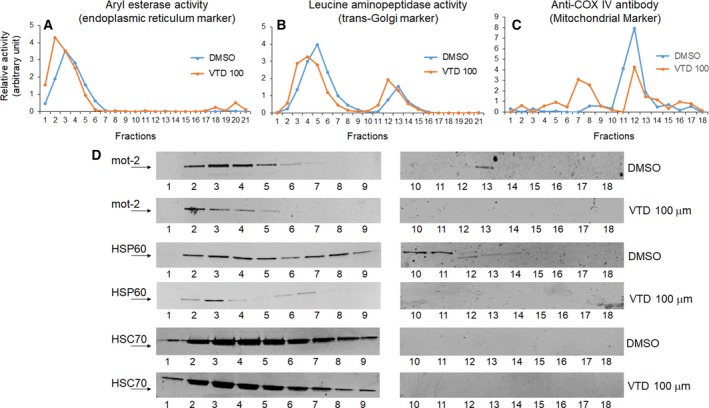
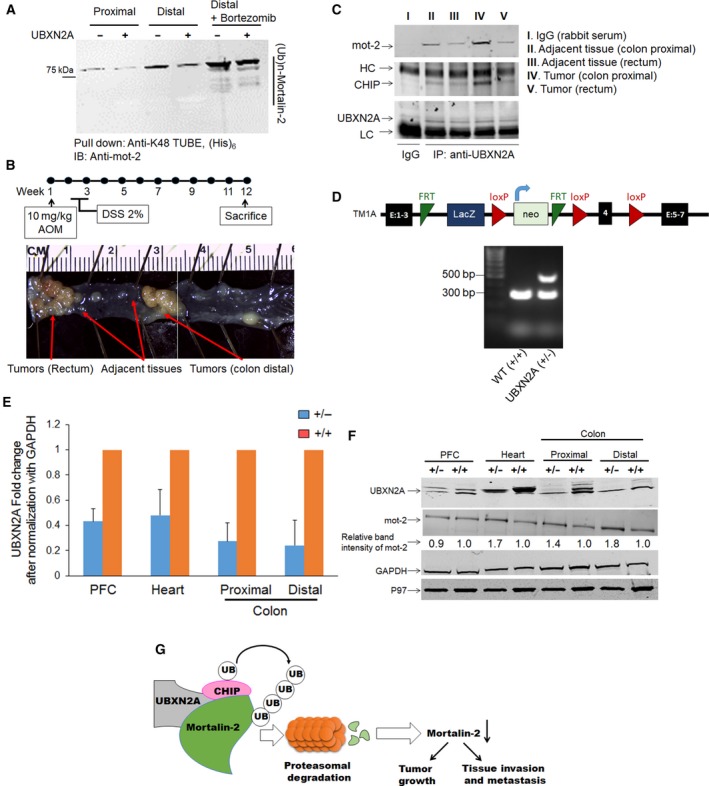
Overexpression of oncoproteins is a major cause of treatment failure using current chemotherapeutic drugs. Drug-induced degradation of oncoproteins is feasible and can improve clinical outcomes in diverse types of cancers. Mortalin-2 (mot-2) is a dominant oncoprotein in several tumors, including colorectal cancer (CRC). In addition to inactivating the p53 tumor suppressor protein, mot-2 enhances tumor cell invasion and migration. Thus, mot-2 is considered a potential therapeutic target in several cancer types. The current study investigated the biological role of a ubiquitin-like protein called UBXN2A in the regulation of mot-2 turnover. An orthogonal ubiquitin transfer technology followed by immunoprecipitation, in vitro ubiquitination, and Magnetic Beads TUBE2 pull-down experiments revealed that UBXN2A promotes carboxyl terminus of the HSP70-interacting protein (CHIP)-dependent ubiquitination of mot-2. We subsequently showed that UBXN2A increases proteasomal degradation of mot-2. A subcellular compartmentalization experiment revealed that induced UBXN2A decreases the level of mot-2 and its chaperone partner, HSP60. Pharmacological upregulation of UBXN2A using a small molecule, veratridine (VTD), decreases the level of mot-2 in cancer cells. Consistent with the in vitro results, UBXN2A+/- mice exhibited selective elevation of mot-2 in colon tissues. An in vitro Anti-K48 TUBE isolation approach showed that recombinant UBXN2A enhances proteasomal degradation of mot-2 in mouse colon tissues. Finally, we observed enhanced association of CHIP with the UBXN2A-mot-2 complex in tumors in an azoxymethane/dextran sulfate sodium-induced mouse CRC model. The existence of a multiprotein complex containing UBXN2A, CHIP, and mot-2 suggests a synergistic tumor suppressor activity of UBXN2A and CHIP in mot-2-enriched tumors. This finding validates the UBXN2A-CHIP axis as a novel and potential therapeutic target in CRC.
Keywords: CHIP E3 ligase; UBXN2A; colorectal cancer; mortalin-2; mouse; veratridine.
© 2018 The Authors. Published by FEBS Press and John Wiley & Sons Ltd.
Figures


Similar articles
-
A plant alkaloid, veratridine, potentiates cancer chemosensitivity by UBXN2A-dependent inhibition of an oncoprotein, mortalin-2.Oncotarget. 2015 Sep 15;6(27):23561-81. doi: 10.18632/oncotarget.4452. Oncotarget. 2015. PMID: 26188124 Free PMC article.
-
Ubiquitin-like (UBX)-domain-containing protein, UBXN2A, promotes cell death by interfering with the p53-Mortalin interactions in colon cancer cells.Cell Death Dis. 2014 Mar 13;5(3):e1118. doi: 10.1038/cddis.2014.100. Cell Death Dis. 2014. PMID: 24625977 Free PMC article.
-
Structural studies of UBXN2A and mortalin interaction and the putative role of silenced UBXN2A in preventing response to chemotherapy.Cell Stress Chaperones. 2016 Mar;21(2):313-26. doi: 10.1007/s12192-015-0661-5. Epub 2015 Dec 4. Cell Stress Chaperones. 2016. PMID: 26634371 Free PMC article.
-
[The role of different E3 ubiquitin ligases in regulation of the P53 tumor suppressor protein].Tsitologiia. 2013;55(10):673-87. Tsitologiia. 2013. PMID: 25509121 Review. Russian.
-
CHIP: a co-chaperone for degradation by the proteasome.Subcell Biochem. 2015;78:219-42. doi: 10.1007/978-3-319-11731-7_11. Subcell Biochem. 2015. PMID: 25487024 Review.
Cited by
-
Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy.Cells. 2019 Dec 24;9(1):60. doi: 10.3390/cells9010060. Cells. 2019. PMID: 31878360 Free PMC article. Review.
-
A fungal core effector exploits the OsPUX8B.2-OsCDC48-6 module to suppress plant immunity.Nat Commun. 2024 Mar 22;15(1):2559. doi: 10.1038/s41467-024-46903-7. Nat Commun. 2024. PMID: 38519521 Free PMC article.
-
Clinicopathologic Significance of Heat Shock Protein 60 as a Survival Predictor in Colorectal Cancer.Cancers (Basel). 2023 Aug 11;15(16):4052. doi: 10.3390/cancers15164052. Cancers (Basel). 2023. PMID: 37627080 Free PMC article.
-
Veratridine, a plant-derived alkaloid, suppresses the hyperactive Rictor-mTORC2 pathway: a new targeted therapy for primary and metastatic colorectal cancer.Res Sq [Preprint]. 2024 Oct 25:rs.3.rs-5199838. doi: 10.21203/rs.3.rs-5199838/v1. Res Sq. 2024. PMID: 39502780 Free PMC article. Preprint.
-
Ubiquitination regulation of mitochondrial homeostasis: a new sight for the treatment of gastrointestinal tumors.Front Immunol. 2025 Mar 11;16:1533007. doi: 10.3389/fimmu.2025.1533007. eCollection 2025. Front Immunol. 2025. PMID: 40134432 Free PMC article. Review.
References
-
- Agsteribbe E, Huckriede A, Veenhuis M, Ruiters MH, Niezen‐Koning KE, Skjeldal OH, Skullerud K, Gupta RS, Hallberg R, van Diggelen OP et al (1993) A fatal, systemic mitochondrial disease with decreased mitochondrial enzyme activities, abnormal ultrastructure of the mitochondria and deficiency of heat shock protein 60. Biochem Biophys Res Commun 193, 146–154. - PubMed
-
- Ando K, Oki E, Zhao Y, Ikawa‐Yoshida A, Kitao H, Saeki H, Kimura Y, Ida S, Morita M, Kusumoto T et al (2013) Mortalin is a prognostic factor of gastric cancer with normal p53 function. Gastric Cancer 17, 255–262. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
