

Integrated Genomic Analysis of Hürthle Cell Cancer Reveals Oncogenic Drivers, Recurrent Mitochondrial Mutations, and Unique Chromosomal Landscapes
- PMID: 30107176
- PMCID: PMC6247912
- DOI: 10.1016/j.ccell.2018.07.002
Integrated Genomic Analysis of Hürthle Cell Cancer Reveals Oncogenic Drivers, Recurrent Mitochondrial Mutations, and Unique Chromosomal Landscapes
Abstract
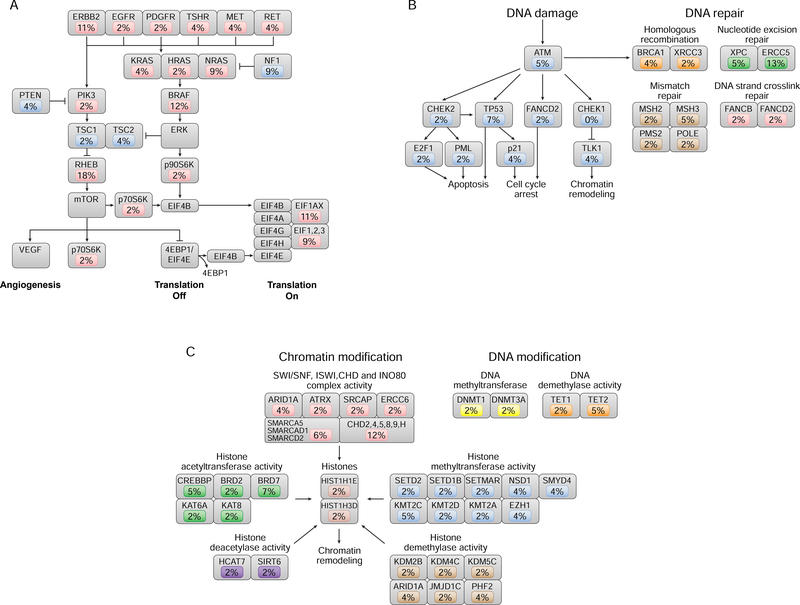
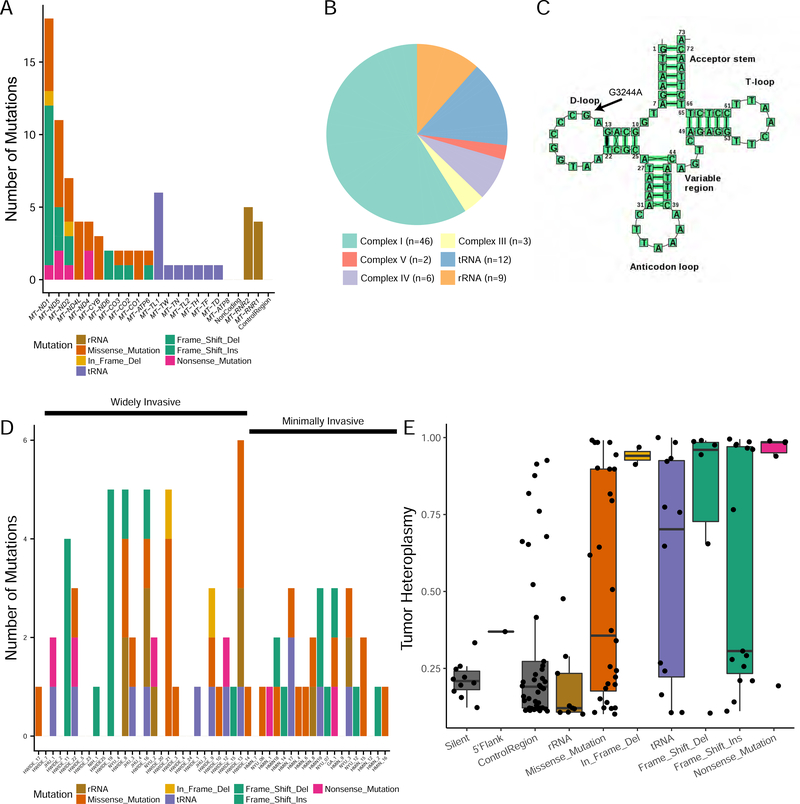
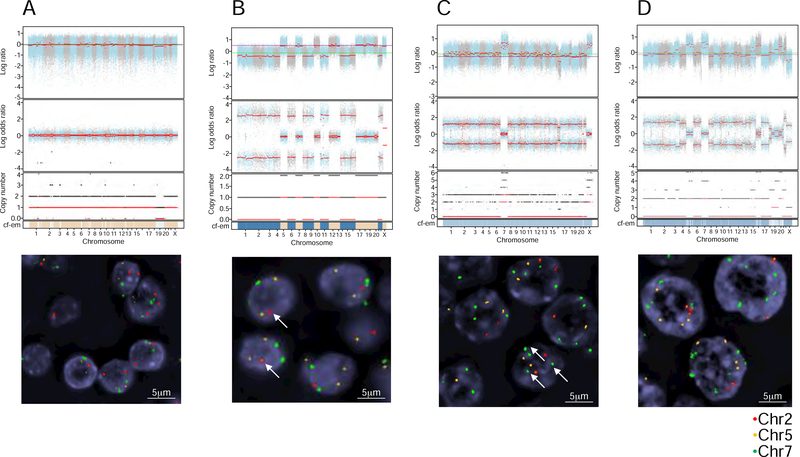
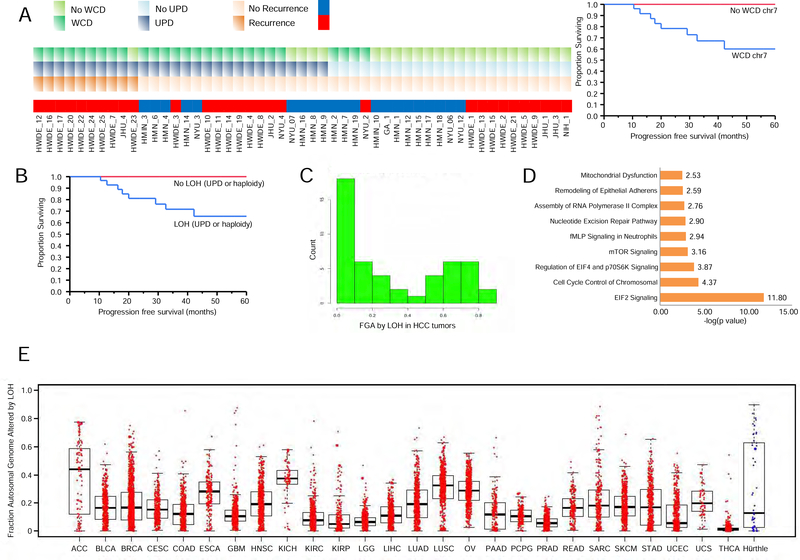
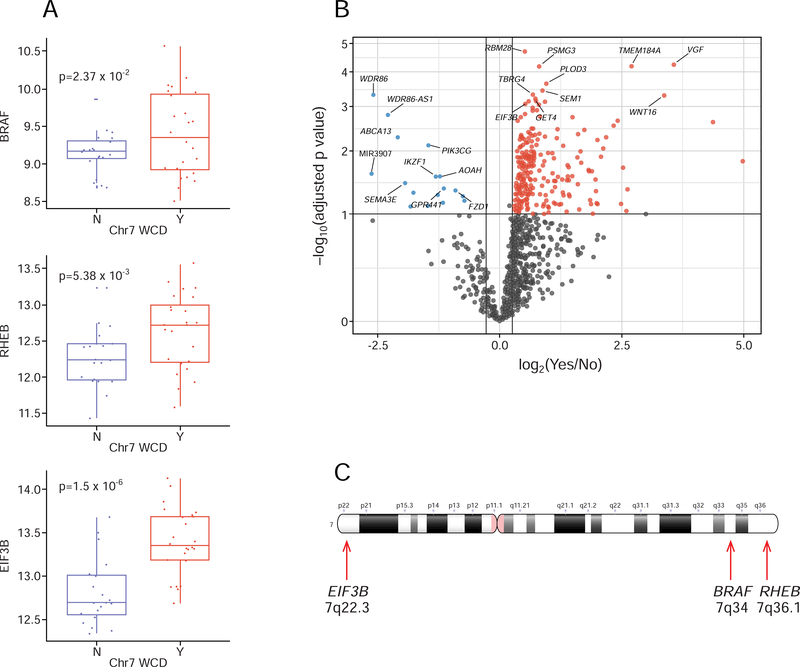
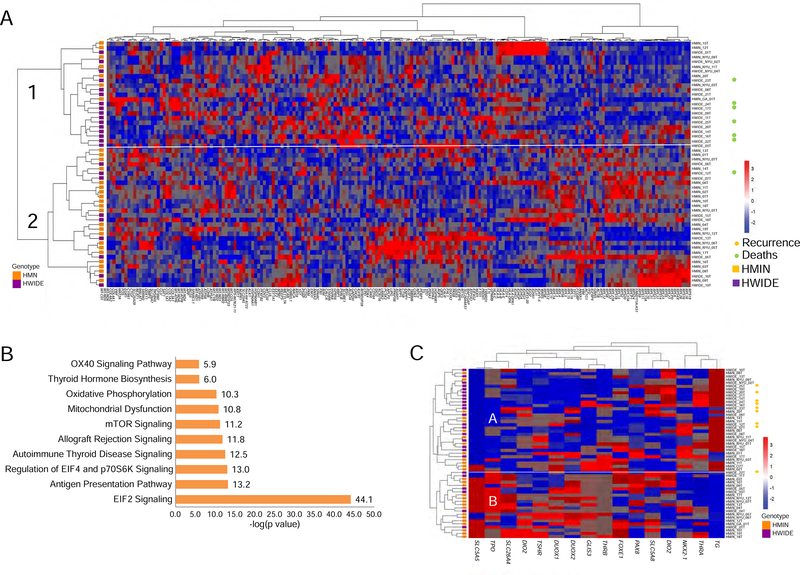
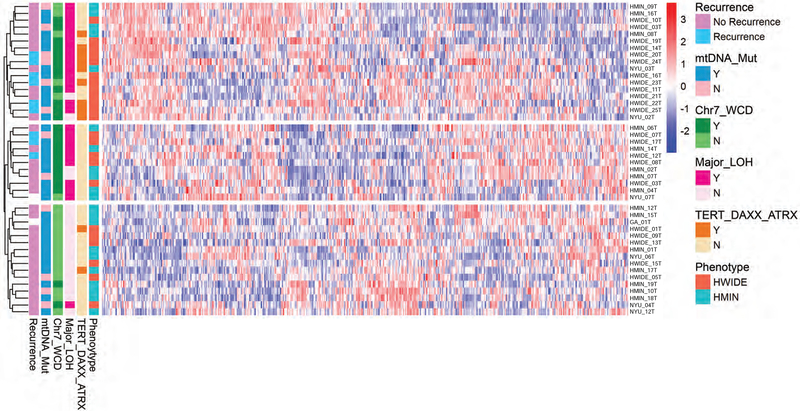
The molecular foundations of Hürthle cell carcinoma (HCC) are poorly understood. Here we describe a comprehensive genomic characterization of 56 primary HCC tumors that span the spectrum of tumor behavior. We elucidate the mutational profile and driver mutations and show that these tumors exhibit a wide range of recurrent mutations. Notably, we report a high number of disruptive mutations to both protein-coding and tRNA-encoding regions of the mitochondrial genome. We reveal unique chromosomal landscapes that involve whole-chromosomal duplications of chromosomes 5 and 7 and widespread loss of heterozygosity arising from haploidization and copy-number-neutral uniparental disomy. We also identify fusion genes and disrupted signaling pathways that may drive disease pathogenesis.
Keywords: Hurthle cell carcinoma; copy-number alterations; fusion genes; genomics; mitochondrial mutations; transcriptome.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures

References
-
- Besic N, Vidergar-Kralj B, Frkovic-Grazio S, Movrin-Stanovnik T, and Auersperg M (2003). The role of radioactive iodine in the treatment of Hürthle cell carcinoma of the thyroid. Thyroid 13, 577–584. - PubMed
-
- Boot A, Oosting J, de Miranda NF, Zhang Y, Corver WE, van de Water B, Morreau H, and van Wezel T (2016). Imprinted survival genes preclude loss of heterozygosity of chromosome 7 in cancer cells. J Pathol 240, 72–83. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
