

The clinical spectrum and genetic variability of limb-girdle muscular dystrophy in a cohort of Chinese patients
- PMID: 30107846
- PMCID: PMC6092860
- DOI: 10.1186/s13023-018-0859-6
The clinical spectrum and genetic variability of limb-girdle muscular dystrophy in a cohort of Chinese patients
Abstract
Background: Limb-girdle muscular dystrophy (LGMD) is a commonly diagnosed hereditary muscular disorder, characterized by the progressive weakness of the limb-girdle muscles. Although the condition has been well-characterized, clinical and genetic heterogeneity can be observed in patients with LGMD. Here, we aimed to describe the clinical manifestations and genetic variability among a cohort of patients with LGMD in South China.
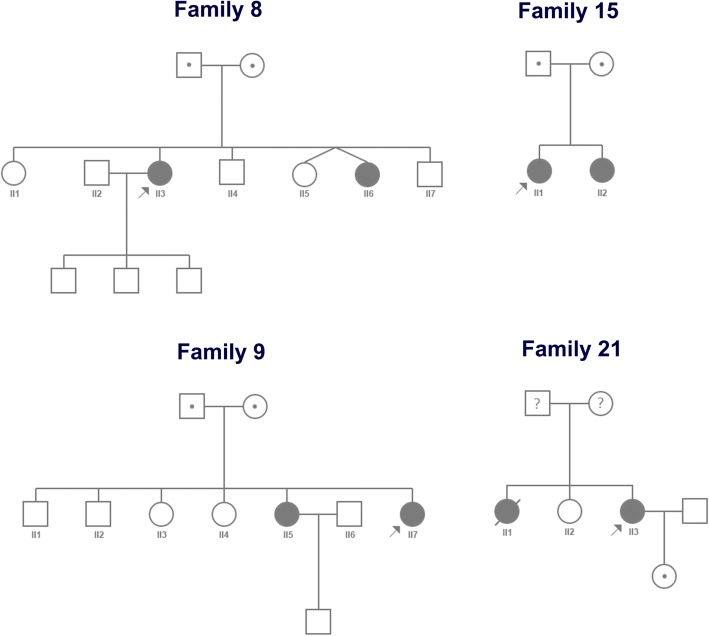
Results: We analyzed the clinical information, muscle magnetic resonance imaging (MRI) findings, and genetic results obtained from 30 patients (24 families) with clinically suspected LGMD. In 24 probands, 38 variants were found in total, of which 18 were shown to be novel. Among the 30 patients, the most common subtypes were dysferlinopathy in eight (26.67%), sarcoglycanopathies in eight [26.67%; LGMD 2C in three (10.00%), LGMD 2D in three (10.00%), and LGMD 2F in two (6.67%)], LGMD 2A in seven (23.33%), followed by LGMD 1B in three (10.00%), LGMD 2I in three (10.00%), and early onset recessive Emery-Dreifuss-like phenotype without cardiomyopathy in one (3.33%). Furthermore, we also observed novel clinical presentations for LGMD 1B, 2F, and 2I patients with hypermobility of the joints in the upper limbs, a LGMD 2F patient with delayed language development, and other manifestations. Moreover, distinct distributions of fatty infiltration in patients with LGMD 2A, dysferlinopathy, and the early onset recessive Emery-Dreifuss-like phenotype without cardiomyopathy were also observed based on muscle MRI results.
Conclusions: In this study, we expanded the clinical spectrum and genetic variability found in patients with LGMD, which provided additional insights into genotype and phenotype correlations in this disease.
Keywords: Clinical manifestation; Limb-girdle muscular dystrophy; Molecular diagnosis; Muscle magnetic resonance imaging; South China.
Conflict of interest statement
Ethics approval and consent to participate
This study and all protocols used conformed to the ethical guidelines of the 1975 Declaration of Helsinki, as reflected in an a priori approval by the Human Research Committee of the Hereditary Neurological Disease Clinics of The First Affiliated Hospital, Sun Yat-sen University. Informed consent was obtained from each patient included in the study.
Consent for publication
The adult patients and the parents of the children described in this article provided consent for participation in the study and for publishing the obtained results.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures


References
-
- Liewluck T, Milone M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018;[Epub ahead of print]. 10.1002/mus.26077. - PubMed
Publication types
MeSH terms
Grants and funding
- 81771359, 81471280 and 81271401/Natural Science Foundation of China/International
- 2014A020212130/Guangdong Provincial Science and Technology Plan/International
- 1561000153(201508020012)/Guangzhou City Science and Technology Plan/International
- 2014B030301035/National Key Clinical Department and Key Discipline of Neurology, the Guangdong Provincial Key Laboratory for Diagnosis and Treatment of Major Neurological Diseases/International
- 2015B050501003/Southern China International Cooperation Base for Early Intervention and Functional Rehabilitation of Neurological Diseases/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
