

Kub5-Hera RPRD1B Deficiency Promotes "BRCAness" and Vulnerability to PARP Inhibition in BRCA-proficient Breast Cancers
- PMID: 30108102
- PMCID: PMC6295248
- DOI: 10.1158/1078-0432.CCR-17-1118
Kub5-Hera RPRD1B Deficiency Promotes "BRCAness" and Vulnerability to PARP Inhibition in BRCA-proficient Breast Cancers
Abstract
Purpose: Identification of novel strategies to expand the use of PARP inhibitors beyond BRCA deficiency is of great interest in personalized medicine. Here, we investigated the unannotated role of Kub5-HeraRPRD1B (K-H) in homologous recombination (HR) repair and its potential clinical significance in targeted cancer therapy.
Experimental design: Functional characterization of K-H alterations on HR repair of double-strand breaks (DSB) were assessed by targeted gene silencing, plasmid reporter assays, immunofluorescence, and Western blots. Cell survival with PARP inhibitors was evaluated through colony-forming assays and statistically analyzed for correlation with K-H expression in various BRCA1/2 nonmutated breast cancers. Gene expression microarray/qPCR analyses, chromatin immunoprecipitation, and rescue experiments were used to investigate molecular mechanisms of action.
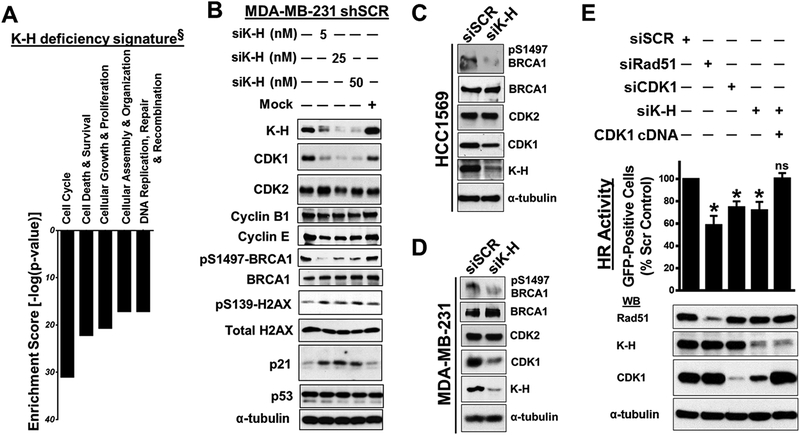
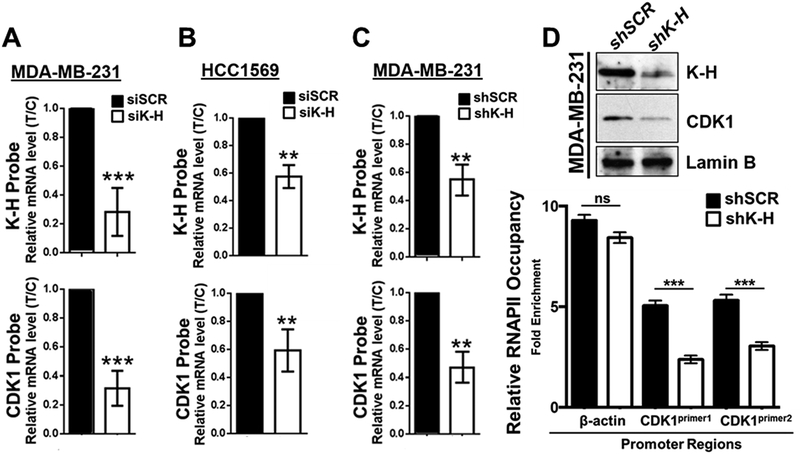
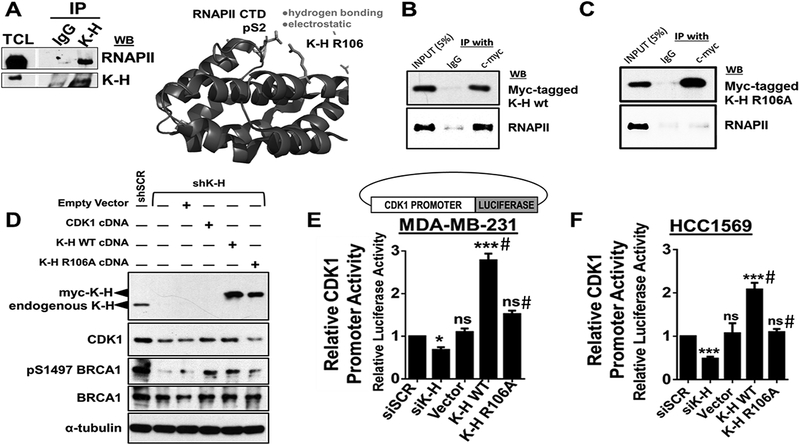
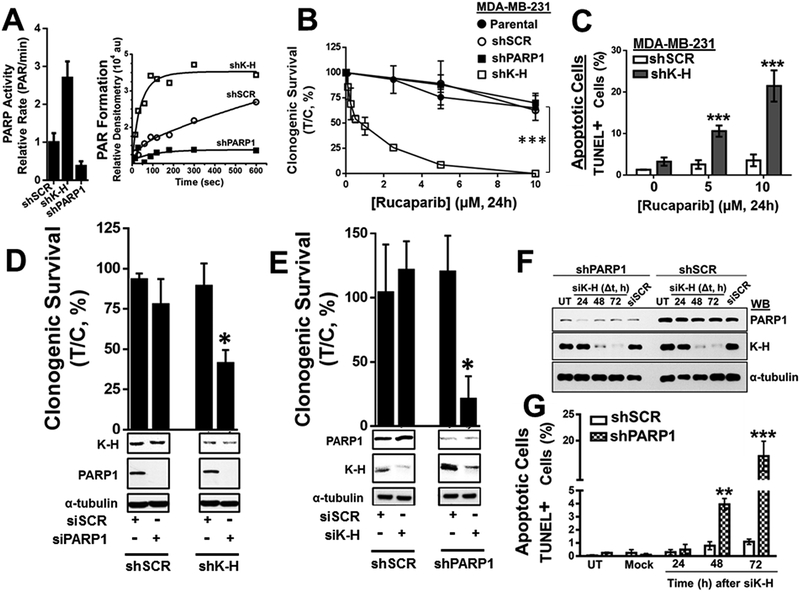
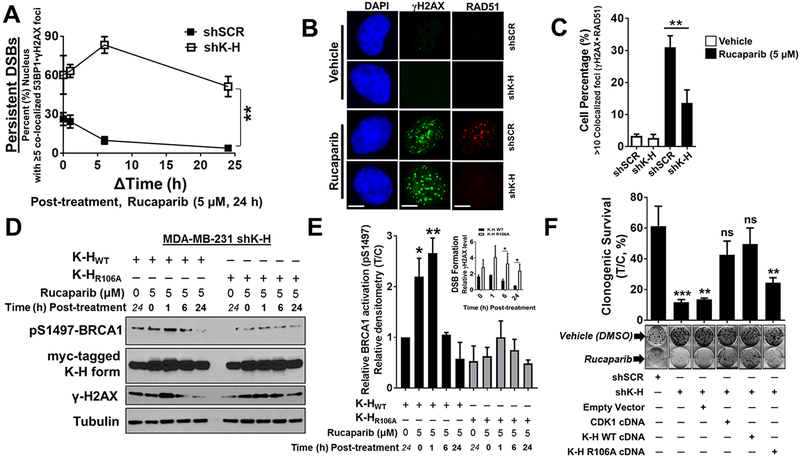
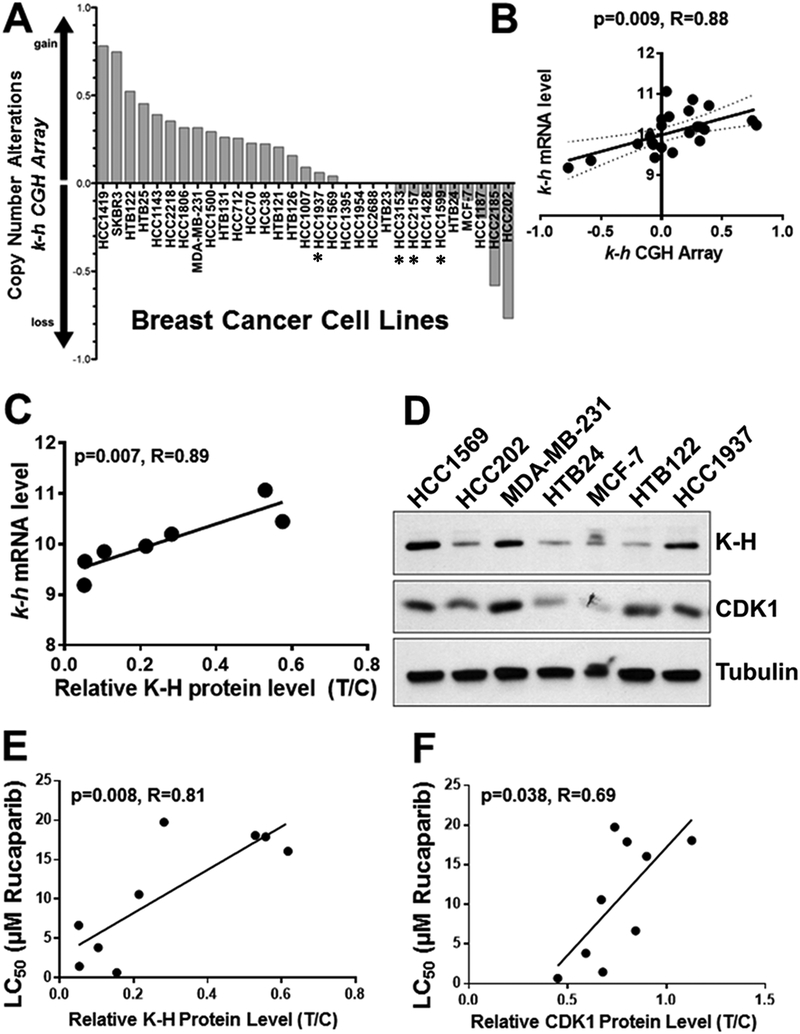
Results: K-H expression loss correlates with rucaparib LD50 values in a panel of BRCA1/2 nonmutated breast cancers. Mechanistically, K-H depletion promotes BRCAness, where extensive upregulation of PARP1 activity was required for the survival of breast cancer cells. PARP inhibition in these cells led to synthetic lethality that was rescued by wild-type K-H reexpression, but not by a mutant K-H (p.R106A) that weakly binds RNAPII. K-H mediates HR by facilitating recruitment of RNAPII to the promoter region of a critical DNA damage response and repair effector, cyclin-dependent kinase 1 (CDK1).
Conclusions: Cancer cells with low K-H expression may have exploitable BRCAness properties that greatly expand the use of PARP inhibitors beyond BRCA mutations. Our results suggest that aberrant K-H alterations may have vital translational implications in cellular responses/survival to DNA damage, carcinogenesis, and personalized medicine.
©2018 American Association for Cancer Research.
Conflict of interest statement
Figures
Similar articles
-
mTOR Inhibitors Suppress Homologous Recombination Repair and Synergize with PARP Inhibitors via Regulating SUV39H1 in BRCA-Proficient Triple-Negative Breast Cancer.Clin Cancer Res. 2016 Apr 1;22(7):1699-712. doi: 10.1158/1078-0432.CCR-15-1772. Epub 2015 Nov 6. Clin Cancer Res. 2016. PMID: 26546619 Free PMC article.
-
PARP inhibitors suppress tumours via centrosome error-induced senescence independent of DNA damage response.EBioMedicine. 2024 May;103:105129. doi: 10.1016/j.ebiom.2024.105129. Epub 2024 Apr 18. EBioMedicine. 2024. PMID: 38640836 Free PMC article.
-
Androgen receptor inhibitor-induced "BRCAness" and PARP inhibition are synthetically lethal for castration-resistant prostate cancer.Sci Signal. 2017 May 23;10(480):eaam7479. doi: 10.1126/scisignal.aam7479. Sci Signal. 2017. PMID: 28536297 Free PMC article.
-
PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside.Cancer. 2018 Jun 15;124(12):2498-2506. doi: 10.1002/cncr.31307. Epub 2018 Apr 16. Cancer. 2018. PMID: 29660759 Free PMC article. Review.
-
PARP inhibitors in older patients with ovarian and breast cancer: Young International Society of Geriatric Oncology review paper.J Geriatr Oncol. 2019 Mar;10(2):337-345. doi: 10.1016/j.jgo.2018.10.008. Epub 2018 Oct 14. J Geriatr Oncol. 2019. PMID: 30333088 Review.
Cited by
-
XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice.Cancers (Basel). 2020 Jul 7;12(7):1821. doi: 10.3390/cancers12071821. Cancers (Basel). 2020. PMID: 32645903 Free PMC article.
-
Therapeutic Strategies and Biomarkers to Modulate PARP Activity for Targeted Cancer Therapy.Cancers (Basel). 2020 Apr 14;12(4):972. doi: 10.3390/cancers12040972. Cancers (Basel). 2020. PMID: 32295316 Free PMC article. Review.
-
XRN2 interactome reveals its synthetic lethal relationship with PARP1 inhibition.Sci Rep. 2020 Aug 28;10(1):14253. doi: 10.1038/s41598-020-71203-7. Sci Rep. 2020. PMID: 32859985 Free PMC article.
-
Augmented Concentration of Isopentyl-Deoxynyboquinone in Tumors Selectively Kills NAD(P)H Quinone Oxidoreductase 1-Positive Cancer Cells through Programmed Necrotic and Apoptotic Mechanisms.Cancers (Basel). 2023 Dec 14;15(24):5844. doi: 10.3390/cancers15245844. Cancers (Basel). 2023. PMID: 38136388 Free PMC article.
-
PCNA inhibition enhances the cytotoxicity of β-lapachone in NQO1-Positive cancer cells by augmentation of oxidative stress-induced DNA damage.Cancer Lett. 2021 Oct 28;519:304-314. doi: 10.1016/j.canlet.2021.07.040. Epub 2021 Jul 27. Cancer Lett. 2021. PMID: 34329742 Free PMC article.
References
-
- Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012;481(7381):287–94 doi 10.1038/nature10760. - DOI - PubMed
-
- Cancer Fojo T., DNA repair mechanisms, and resistance to chemotherapy. J Natl Cancer Inst 2001;93(19):1434–6. - PubMed
-
- Morales JC, Richard P, Rommel A, Fattah FJ, Motea EA, Patidar PL, et al. Kub5-Hera, the human Rtt103 homolog, plays dual functional roles in transcription termination and DNA repair. Nucleic acids research 2014;42(8):4996–5006 doi 10.1093/nar/gku160. - DOI - PMC - PubMed
-
- Lu D, Wu Y, Wang Y, Ren F, Wang D, Su F, et al. CREPT accelerates tumorigenesis by regulating the transcription of cell-cycle-related genes. Cancer cell 2012;21(1):92–104 doi 10.1016/j.ccr.2011.12.016. - DOI - PubMed
-
- Patidar PL, Motea EA, Fattah FJ, Zhou Y, Morales JC, Xie Y, et al. The Kub5-Hera/RPRD1B interactome: a novel role in preserving genetic stability by regulating DNA mismatch repair. Nucleic acids research 2016;44(4):1718–31 doi 10.1093/nar/gkv1492. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
