

Distinct patterns of histone acetyltransferase and Mediator deployment at yeast protein-coding genes
- PMID: 30108132
- PMCID: PMC6120713
- DOI: 10.1101/gad.312173.118
Distinct patterns of histone acetyltransferase and Mediator deployment at yeast protein-coding genes
Abstract
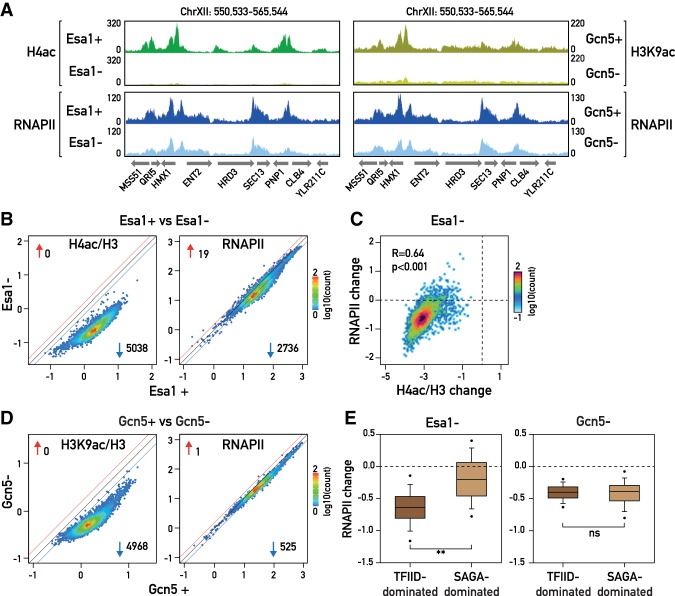
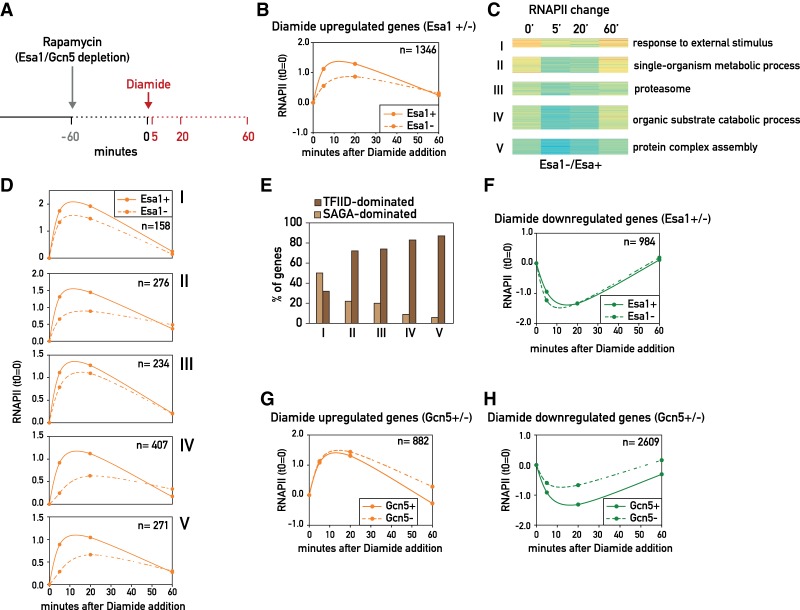
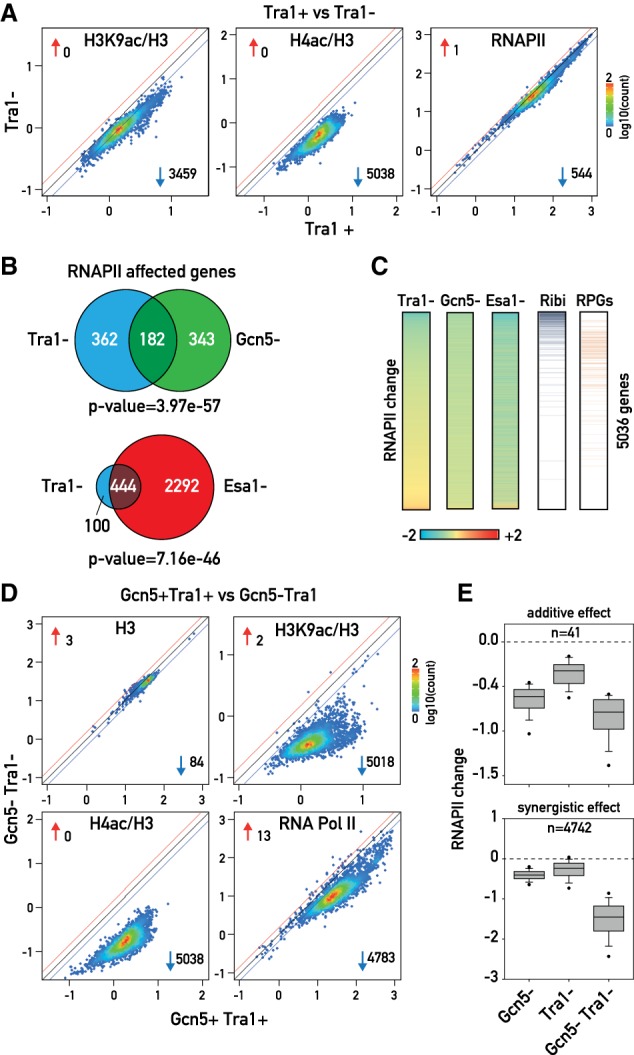
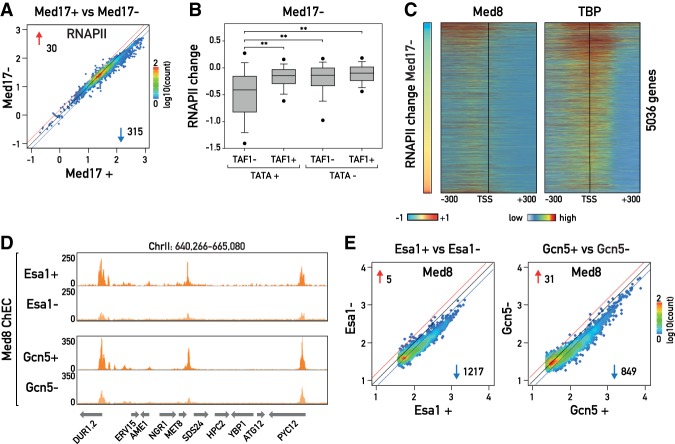
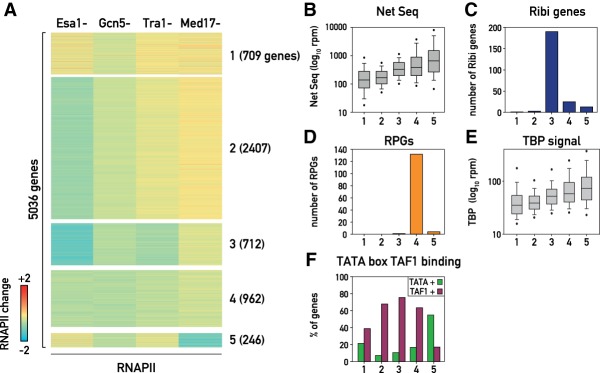
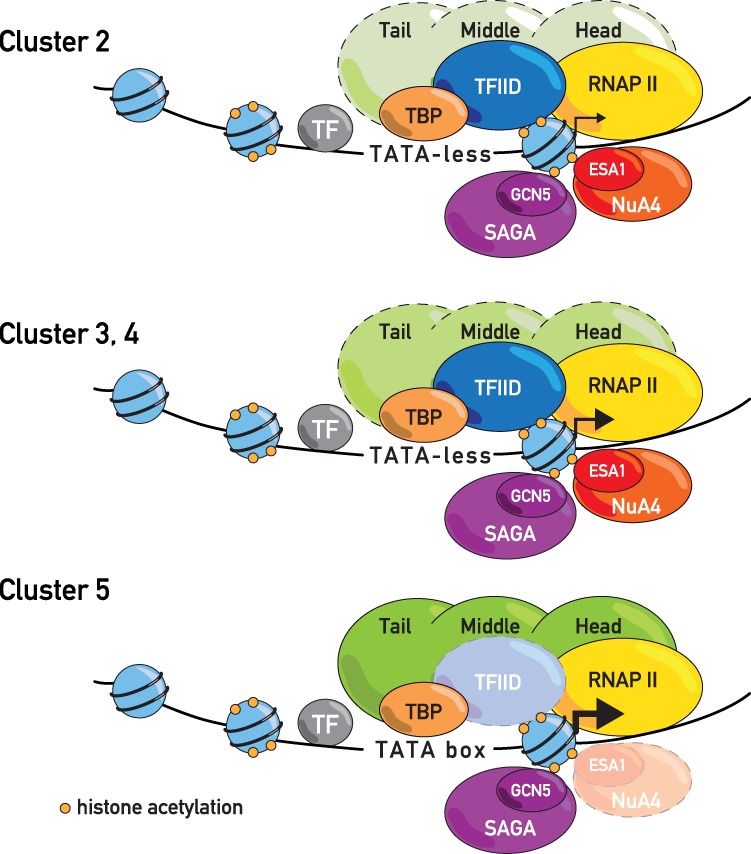
The transcriptional coactivators Mediator and two histone acetyltransferase (HAT) complexes, NuA4 and SAGA, play global roles in transcriptional activation. Here we explore the relative contributions of these factors to RNA polymerase II association at specific genes and gene classes by rapid nuclear depletion of key complex subunits. We show that the NuA4 HAT Esa1 differentially affects certain groups of genes, whereas the SAGA HAT Gcn5 has a weaker but more uniform effect. Relative dependence on Esa1 and Tra1, a shared component of NuA4 and SAGA, distinguishes two large groups of coregulated growth-promoting genes. In contrast, we show that the activity of Mediator is particularly important at a separate, small set of highly transcribed TATA-box-containing genes. Our analysis indicates that at least three distinct combinations of coactivator deployment are used to generate moderate or high transcription levels and suggests that each may be associated with distinct forms of regulation.
Keywords: Esa1; Gcn5; Mediator; Tra1; histone acetylation; transcription.
© 2018 Bruzzone et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Simultaneous recruitment of coactivators by Gcn4p stimulates multiple steps of transcription in vivo.Mol Cell Biol. 2005 Jul;25(13):5626-38. doi: 10.1128/MCB.25.13.5626-5638.2005. Mol Cell Biol. 2005. PMID: 15964818 Free PMC article.
-
NuA4 lysine acetyltransferase Esa1 is targeted to coding regions and stimulates transcription elongation with Gcn5.Mol Cell Biol. 2009 Dec;29(24):6473-87. doi: 10.1128/MCB.01033-09. Epub 2009 Oct 12. Mol Cell Biol. 2009. PMID: 19822662 Free PMC article.
-
SAGA subunits Spt3 and Spt8 act directly and non-redundantly with TFIID in TBP recruitment in the Gcn4 transcriptome.Nucleic Acids Res. 2025 Jul 8;53(13):gkaf598. doi: 10.1093/nar/gkaf598. Nucleic Acids Res. 2025. PMID: 40637224 Free PMC article.
-
Share and share alike: the role of Tra1 from the SAGA and NuA4 coactivator complexes.Transcription. 2019 Feb;10(1):37-43. doi: 10.1080/21541264.2018.1530936. Epub 2018 Oct 30. Transcription. 2019. PMID: 30375921 Free PMC article. Review.
-
Acetylation of histones and transcription-related factors.Microbiol Mol Biol Rev. 2000 Jun;64(2):435-59. doi: 10.1128/MMBR.64.2.435-459.2000. Microbiol Mol Biol Rev. 2000. PMID: 10839822 Free PMC article. Review.
Cited by
-
Dynamic modules of the coactivator SAGA in eukaryotic transcription.Exp Mol Med. 2020 Jul;52(7):991-1003. doi: 10.1038/s12276-020-0463-4. Epub 2020 Jul 3. Exp Mol Med. 2020. PMID: 32616828 Free PMC article. Review.
-
The structure of the NuA4-Tip60 complex reveals the mechanism and importance of long-range chromatin modification.Nat Struct Mol Biol. 2023 Sep;30(9):1337-1345. doi: 10.1038/s41594-023-01056-x. Epub 2023 Aug 7. Nat Struct Mol Biol. 2023. PMID: 37550452
-
NuA4 and H2A.Z control environmental responses and autotrophic growth in Arabidopsis.Nat Commun. 2022 Jan 12;13(1):277. doi: 10.1038/s41467-021-27882-5. Nat Commun. 2022. PMID: 35022409 Free PMC article.
-
The SAGA and NuA4 component Tra1 regulates Candida albicans drug resistance and pathogenesis.Genetics. 2021 Oct 2;219(2):iyab131. doi: 10.1093/genetics/iyab131. Genetics. 2021. PMID: 34849885 Free PMC article.
-
SETD2: from chromatin modifier to multipronged regulator of the genome and beyond.Cell Mol Life Sci. 2022 Jun 6;79(6):346. doi: 10.1007/s00018-022-04352-9. Cell Mol Life Sci. 2022. PMID: 35661267 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous