

Developmental Dynamics of Long Noncoding RNA Expression during Sexual Fruiting Body Formation in Fusarium graminearum
- PMID: 30108170
- PMCID: PMC6094484
- DOI: 10.1128/mBio.01292-18
Developmental Dynamics of Long Noncoding RNA Expression during Sexual Fruiting Body Formation in Fusarium graminearum
Abstract
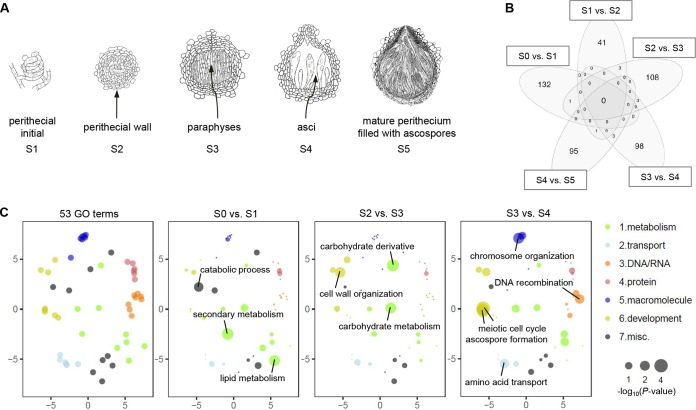
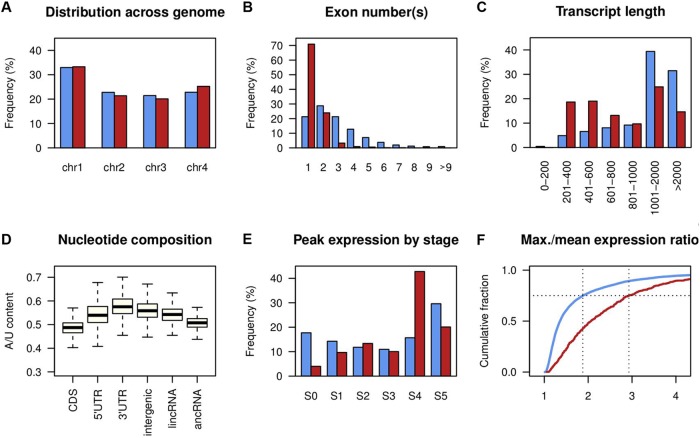
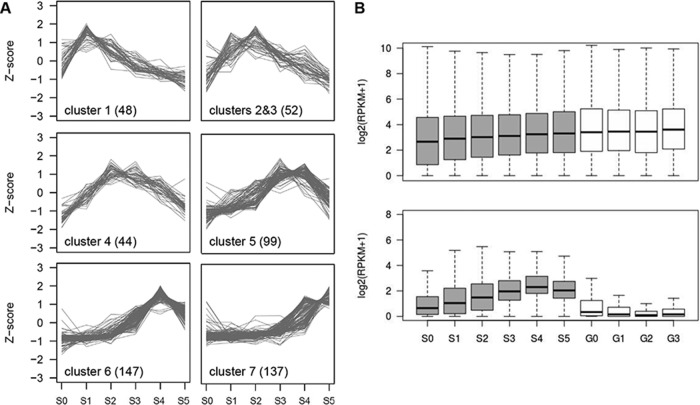
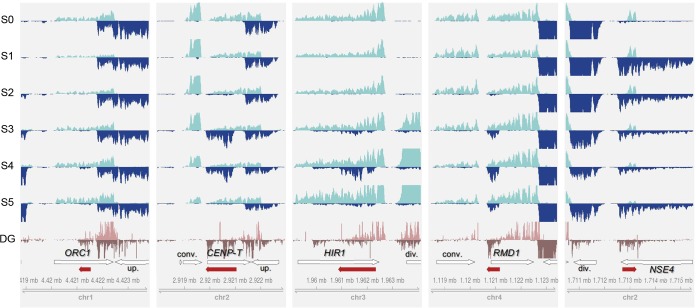
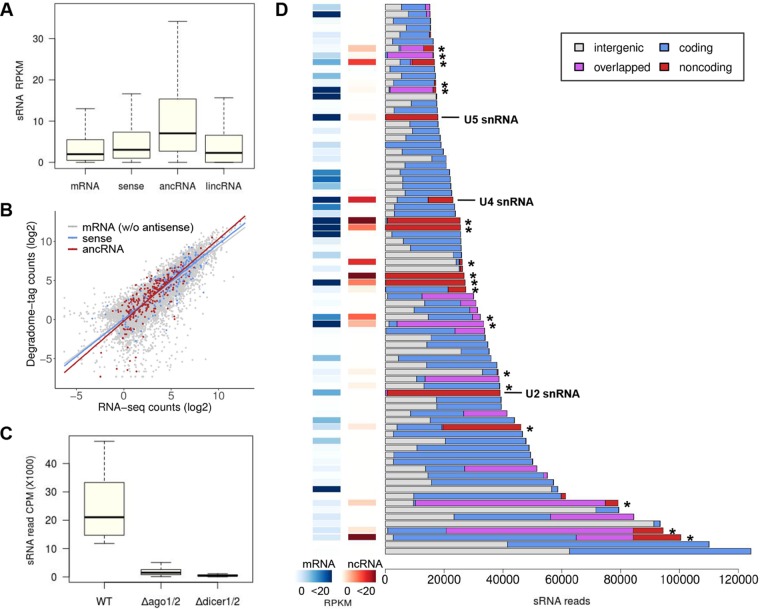
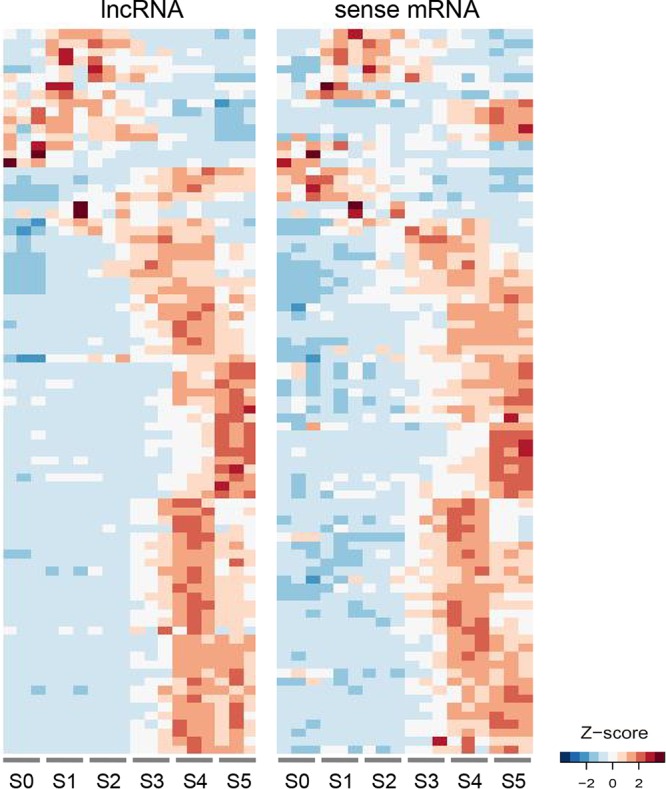
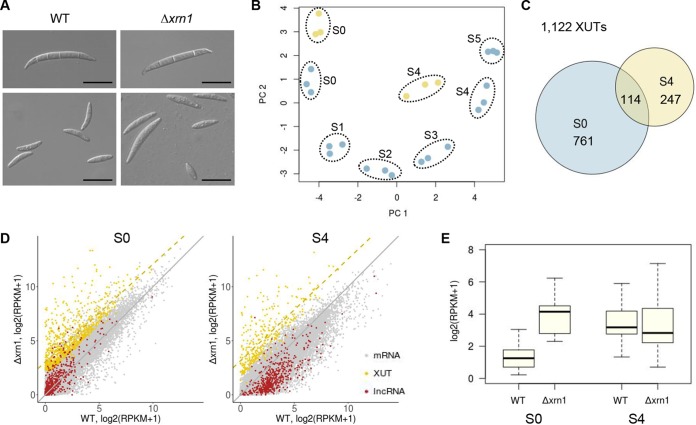
Long noncoding RNA (lncRNA) plays important roles in sexual development in eukaryotes. In filamentous fungi, however, little is known about the expression and roles of lncRNAs during fruiting body formation. By profiling developmental transcriptomes during the life cycle of the plant-pathogenic fungus Fusarium graminearum, we identified 547 lncRNAs whose expression was highly dynamic, with about 40% peaking at the meiotic stage. Many lncRNAs were found to be antisense to mRNAs, forming 300 sense-antisense pairs. Although small RNAs were produced from these overlapping loci, antisense lncRNAs appeared not to be involved in gene silencing pathways. Genome-wide analysis of small RNA clusters identified many silenced loci at the meiotic stage. However, we found transcriptionally active small RNA clusters, many of which were associated with lncRNAs. Also, we observed that many antisense lncRNAs and their respective sense transcripts were induced in parallel as the fruiting bodies matured. The nonsense-mediated decay (NMD) pathway is known to determine the fates of lncRNAs as well as mRNAs. Thus, we analyzed mutants defective in NMD and identified a subset of lncRNAs that were induced during sexual development but suppressed by NMD during vegetative growth. These results highlight the developmental stage-specific nature and functional potential of lncRNA expression in shaping the fungal fruiting bodies and provide fundamental resources for studying sexual stage-induced lncRNAs.IMPORTANCEFusarium graminearum is the causal agent of the head blight on our major staple crops, wheat and corn. The fruiting body formation on the host plants is indispensable for the disease cycle and epidemics. Long noncoding RNA (lncRNA) molecules are emerging as key regulatory components for sexual development in animals and plants. To date, however, there is a paucity of information on the roles of lncRNAs in fungal fruiting body formation. Here we characterized hundreds of lncRNAs that exhibited developmental stage-specific expression patterns during fruiting body formation. Also, we discovered that many lncRNAs were induced in parallel with their overlapping transcripts on the opposite DNA strand during sexual development. Finally, we found a subset of lncRNAs that were regulated by an RNA surveillance system during vegetative growth. This research provides fundamental genomic resources that will spur further investigations on lncRNAs that may play important roles in shaping fungal fruiting bodies.
Keywords: Fusarium graminearum; Xrn1 exonuclease; fruiting body; long noncoding RNAs; perithecia; sexual development.
Copyright © 2018 Kim et al.
Figures
References
-
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. 2009. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223–227. doi: 10.1038/nature07672. - DOI - PMC - PubMed
-
- Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermüller J, Hofacker IL, Bell I, Cheung E, Drenkow J, Dumais E, Patel S, Helt G, Ganesh M, Ghosh S, Piccolboni A, Sementchenko V, Tammana H, Gingeras TR. 2007. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316:1484–1488. doi: 10.1126/science.1138341. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
