

The pro-oxidant adaptor p66SHC promotes B cell mitophagy by disrupting mitochondrial integrity and recruiting LC3-II
- PMID: 30109811
- PMCID: PMC6984773
- DOI: 10.1080/15548627.2018.1505153
The pro-oxidant adaptor p66SHC promotes B cell mitophagy by disrupting mitochondrial integrity and recruiting LC3-II
Abstract
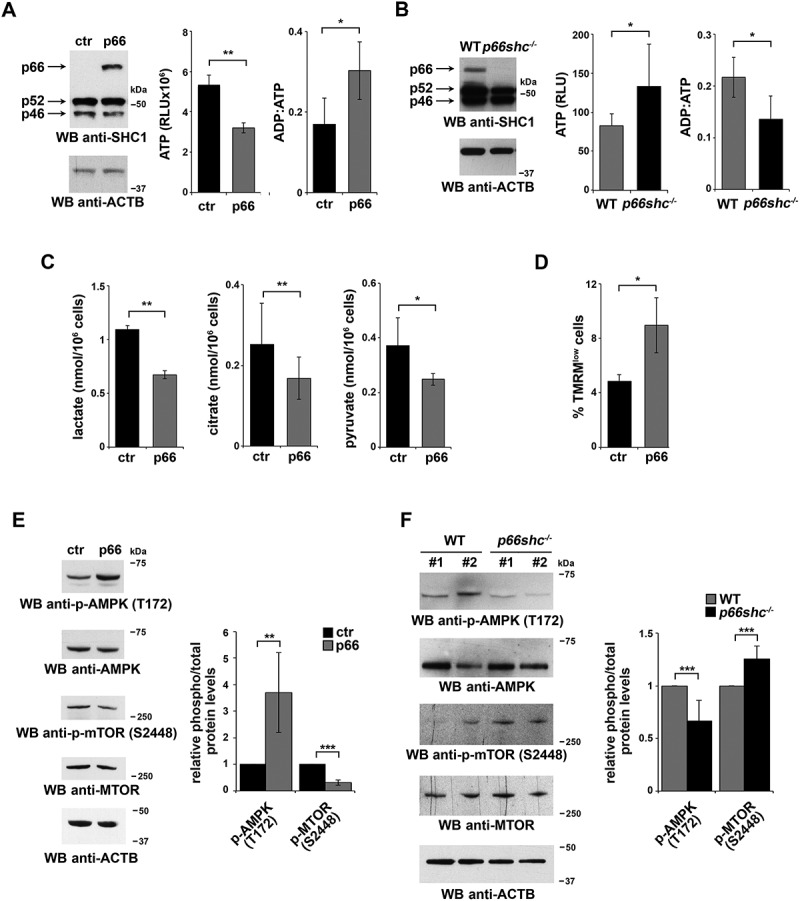
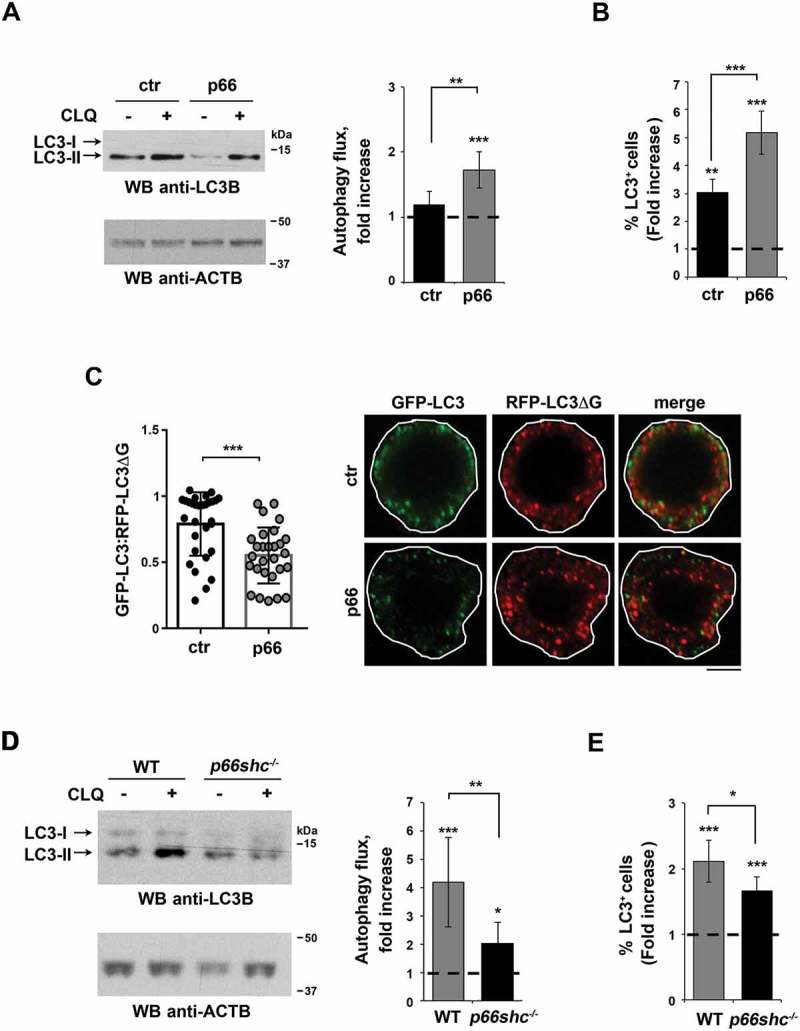
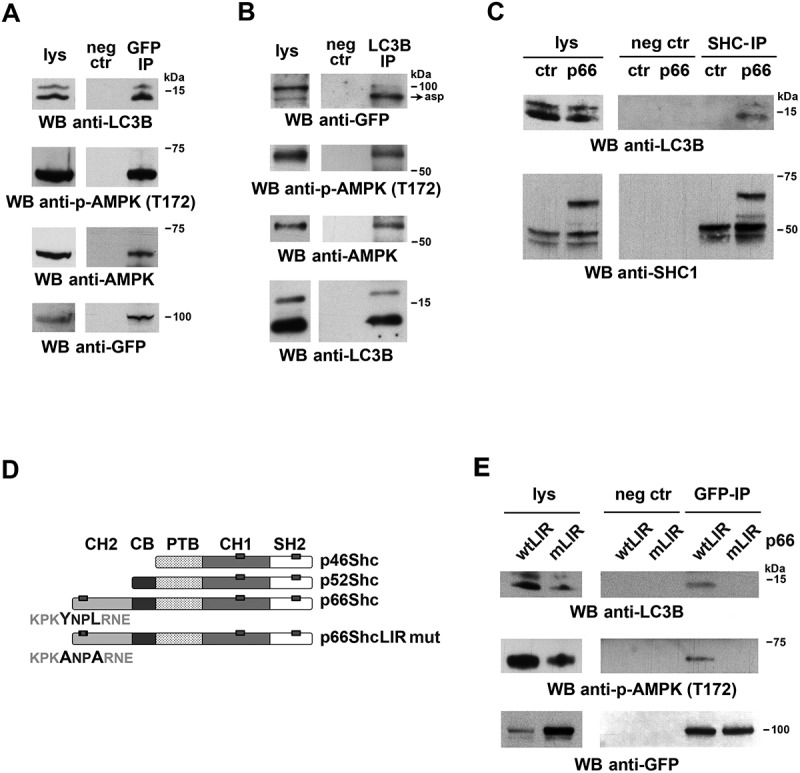
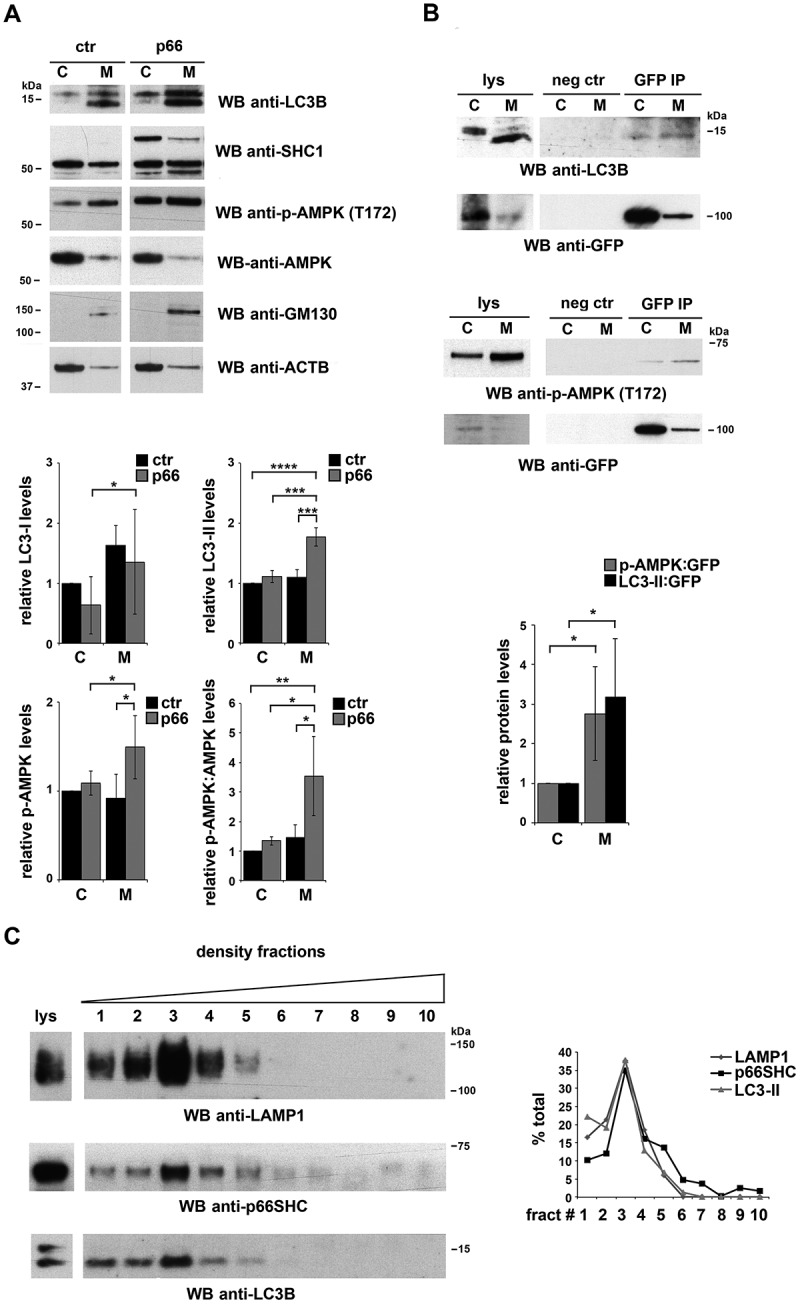
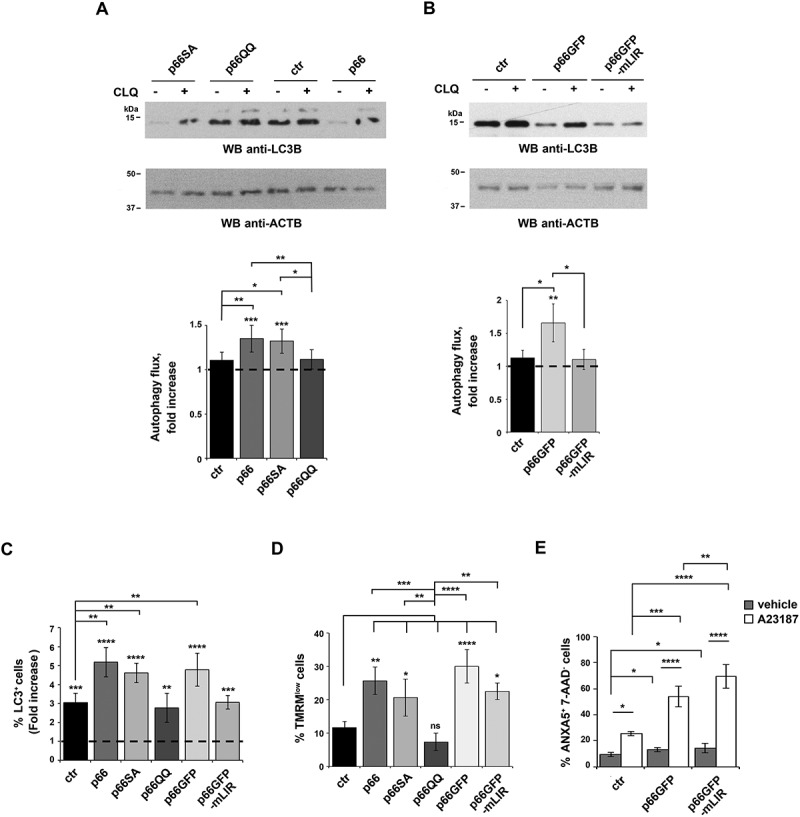
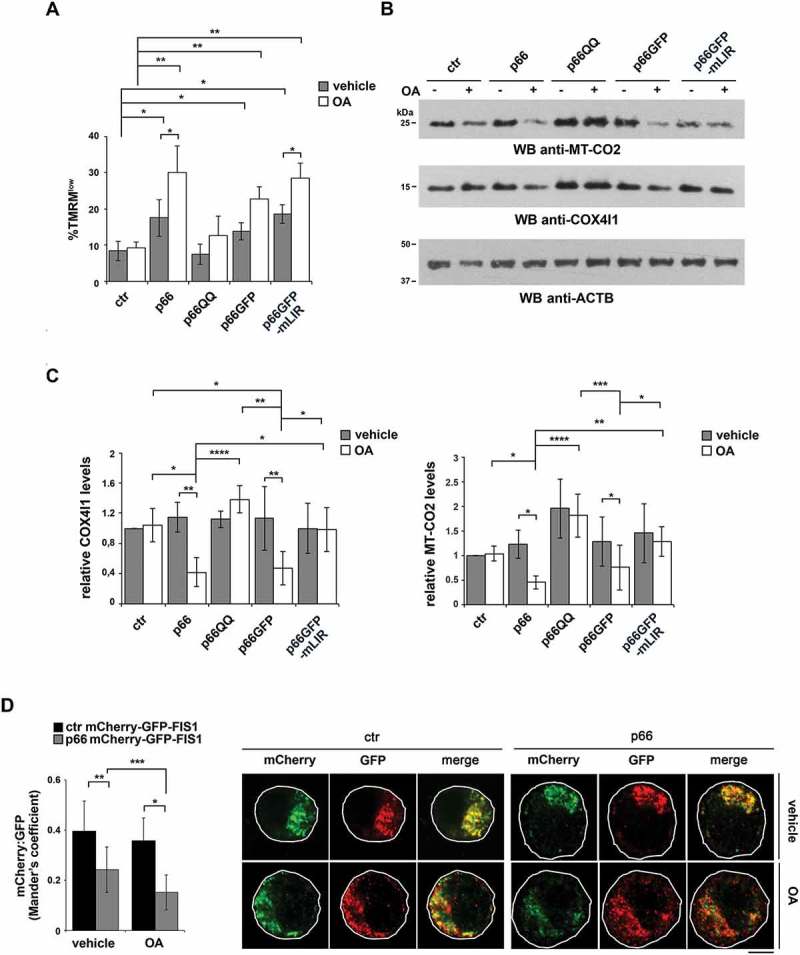
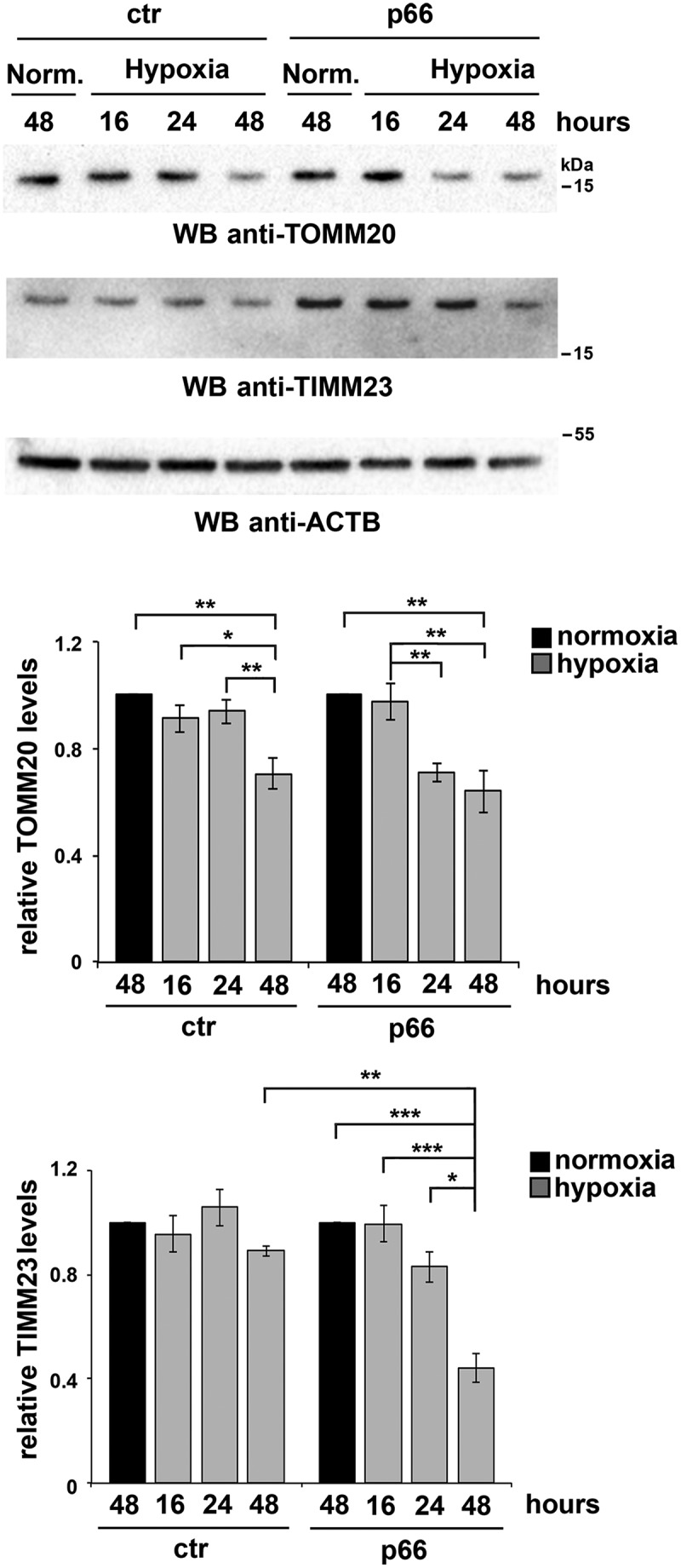
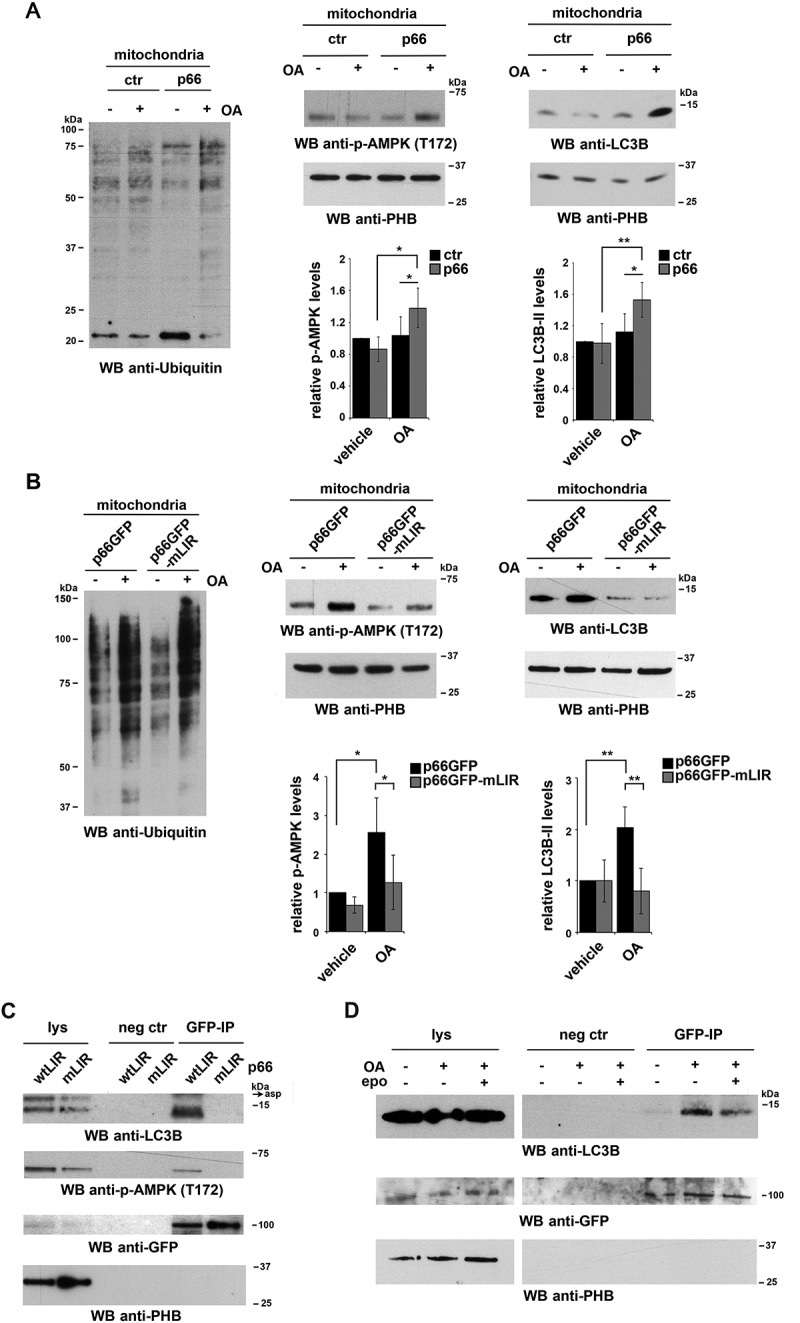
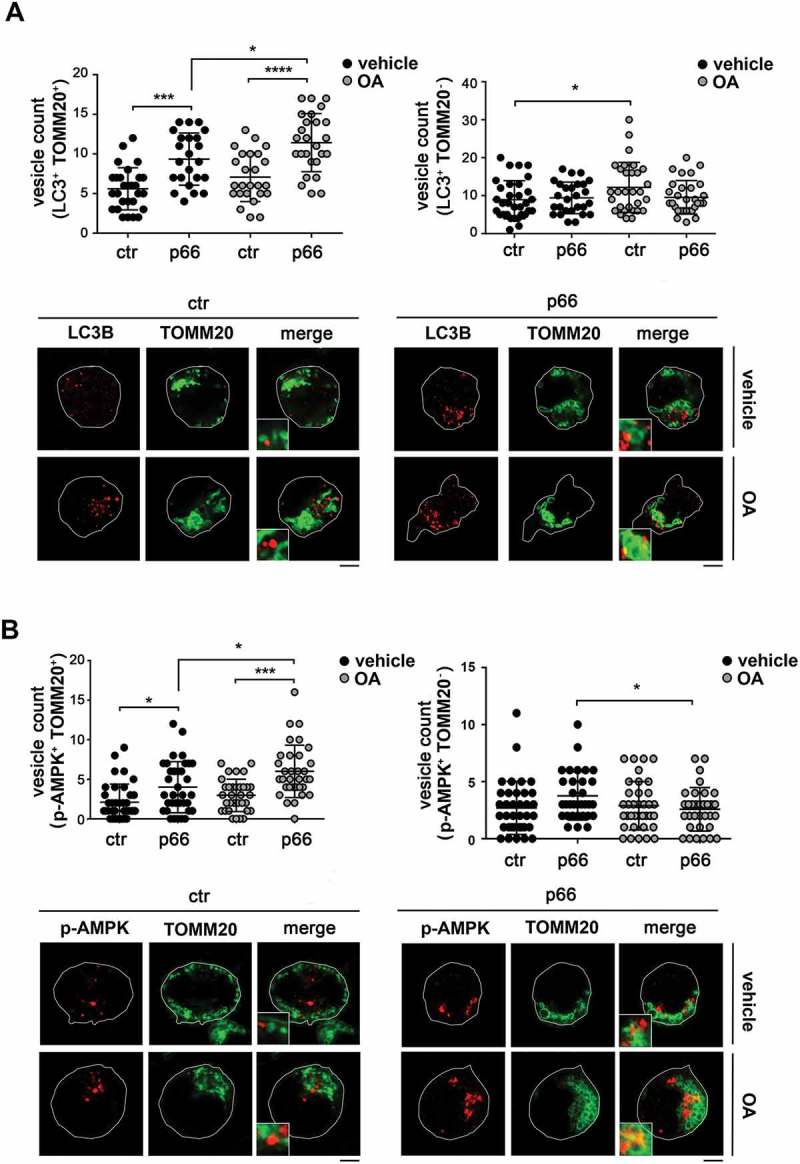
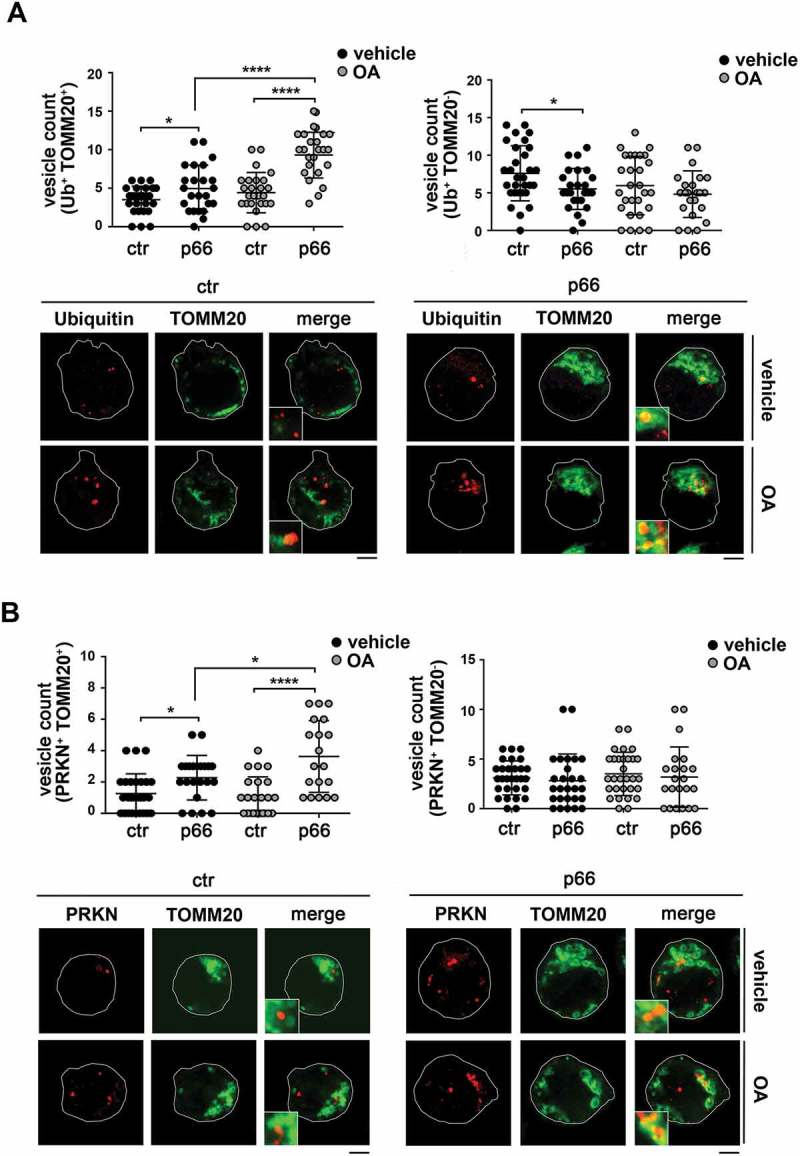
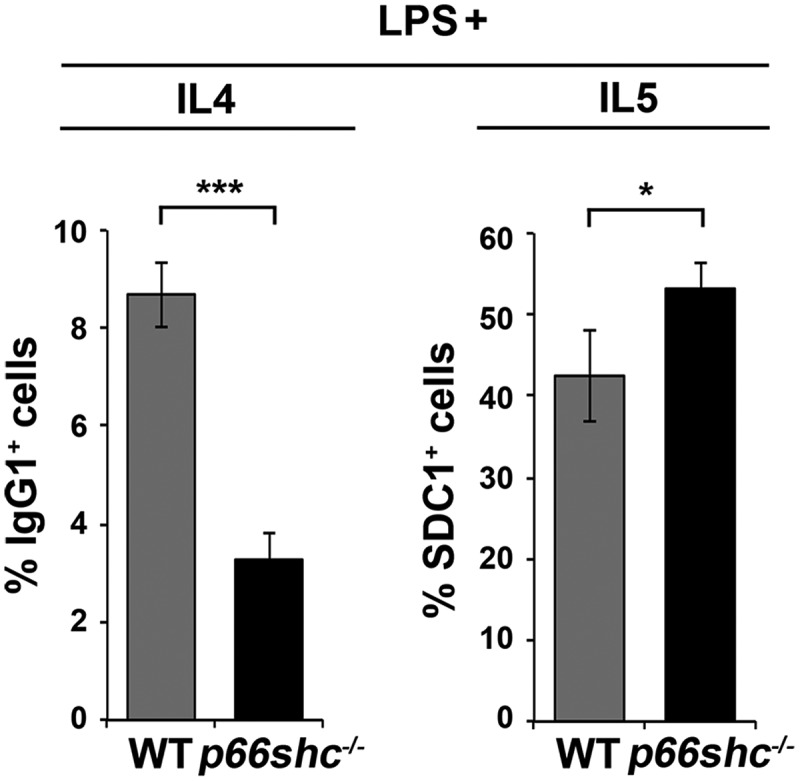
Macroautophagy/autophagy has emerged as a central process in lymphocyte homeostasis, activation and differentiation. Based on our finding that the p66 isoform of SHC1 (p66SHC) pro-apoptotic ROS-elevating SHC family adaptor inhibits MTOR signaling in these cells, here we investigated the role of p66SHC in B-cell autophagy. We show that p66SHC disrupts mitochondrial function through its CYCS (cytochrome c, somatic) binding domain, thereby impairing ATP production, which results in AMPK activation and enhanced autophagic flux. While p66SHC binding to CYCS is sufficient for triggering apoptosis, p66SHC-mediated autophagy additionally depends on its ability to interact with membrane-associated LC3-II through a specific binding motif within its N terminus. Importantly, p66SHC also has an impact on mitochondria homeostasis by inducing mitochondrial depolarization, protein ubiquitination at the outer mitochondrial membrane, and local recruitment of active AMPK. These events initiate mitophagy, whose full execution relies on the role of p66SHC as an LC3-II receptor which brings phagophore membranes to mitochondria. Importantly, p66SHC also promotes hypoxia-induced mitophagy in B cells. Moreover, p66SHC deficiency enhances B cell differentiation to plasma cells, which is controlled by intracellular ROS levels and the hypoxic germinal center environment. The results identify mitochondrial p66SHC as a novel regulator of autophagy and mitophagy in B cells and implicate p66SHC-mediated coordination of autophagy and apoptosis in B cell survival and differentiation. Abbreviations: ACTB: actin beta; AMPK: AMP-activated protein kinase; ATP: adenosine triphosphate; ATG: autophagy-related; CYCS: cytochrome c, somatic; CLQ: chloroquine; COX: cyclooxygenase; CTR: control; GFP: green fluorescent protein; HIFIA/Hif alpha: hypoxia inducible factor 1 subunit alpha; IMS: intermembrane space; LIR: LC3 interacting region; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MTOR/mTOR: mechanistic target of rapamycin kinase; OA: oligomycin and antimycin A; OMM: outer mitochondrial membrane; PHB: prohibitin; PBS: phosphate-buffered saline; PINK1: PTEN induced putative kinase 1; RFP: red fluorescent protein; ROS: reactive oxygen species; SHC: src Homology 2 domain-containing transforming protein; TMRM: tetramethylrhodamine, methyl ester; TOMM: translocase of outer mitochondrial membrane; ULK1: unc-51 like autophagy activating kinase 1; WT: wild-type.
Keywords: Autophagy; B lymphocytes; LC3 adaptor; mitochondria; mitophagy; p66SHC.
Figures
Similar articles
-
Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation.Autophagy. 2019 Sep;15(9):1606-1619. doi: 10.1080/15548627.2019.1591672. Epub 2019 Apr 7. Autophagy. 2019. PMID: 30859901 Free PMC article.
-
Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys.Autophagy. 2019 Dec;15(12):2142-2162. doi: 10.1080/15548627.2019.1615822. Epub 2019 May 22. Autophagy. 2019. PMID: 31066324 Free PMC article.
-
Emerging views of mitophagy in immunity and autoimmune diseases.Autophagy. 2020 Jan;16(1):3-17. doi: 10.1080/15548627.2019.1603547. Epub 2019 Apr 21. Autophagy. 2020. PMID: 30951392 Free PMC article.
-
Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles.Autophagy. 2021 Feb;17(2):385-401. doi: 10.1080/15548627.2020.1725377. Epub 2020 Feb 12. Autophagy. 2021. PMID: 32048886 Free PMC article. Review.
-
Role of AMBRA1 in mitophagy regulation: emerging evidence in aging-related diseases.Autophagy. 2024 Dec;20(12):2602-2615. doi: 10.1080/15548627.2024.2389474. Epub 2024 Sep 2. Autophagy. 2024. PMID: 39113560 Free PMC article. Review.
Cited by
-
Kaempferol Protects Cell Damage in In Vitro Ischemia Reperfusion Model in Rat Neuronal PC12 Cells.Biomed Res Int. 2020 Apr 23;2020:2461079. doi: 10.1155/2020/2461079. eCollection 2020. Biomed Res Int. 2020. PMID: 32382538 Free PMC article.
-
Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1.Autophagy. 2021 Jan;17(1):1-382. doi: 10.1080/15548627.2020.1797280. Epub 2021 Feb 8. Autophagy. 2021. PMID: 33634751 Free PMC article.
-
Low P66shc with High SerpinB3 Levels Favors Necroptosis and Better Survival in Hepatocellular Carcinoma.Biology (Basel). 2021 Apr 23;10(5):363. doi: 10.3390/biology10050363. Biology (Basel). 2021. PMID: 33922660 Free PMC article.
-
Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions.Signal Transduct Target Ther. 2023 Feb 17;8(1):70. doi: 10.1038/s41392-023-01332-8. Signal Transduct Target Ther. 2023. PMID: 36797231 Free PMC article. Review.
-
The role of radiotherapy-related autophagy genes in the prognosis and immune infiltration in lung adenocarcinoma.Front Immunol. 2022 Oct 13;13:992626. doi: 10.3389/fimmu.2022.992626. eCollection 2022. Front Immunol. 2022. PMID: 36311724 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous