

Mechanism of dysfunction of human variants of the IRAK4 kinase and a role for its kinase activity in interleukin-1 receptor signaling
- PMID: 30115681
- PMCID: PMC6166721
- DOI: 10.1074/jbc.RA118.003831
Mechanism of dysfunction of human variants of the IRAK4 kinase and a role for its kinase activity in interleukin-1 receptor signaling
Abstract
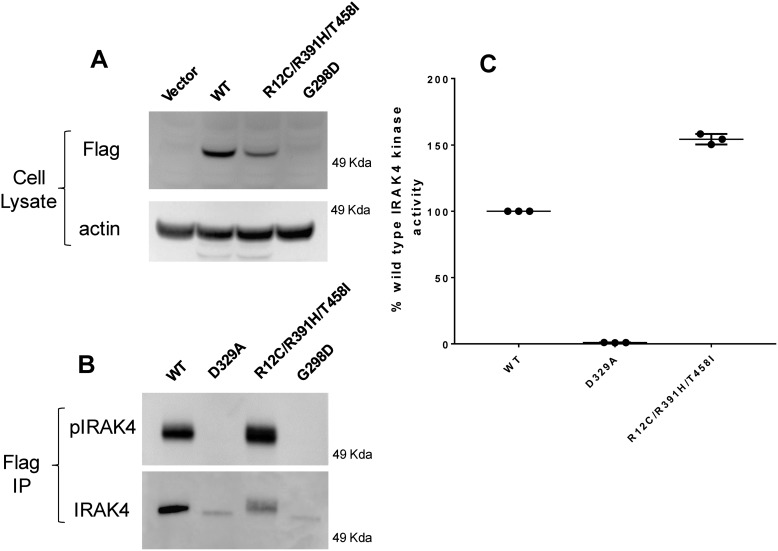
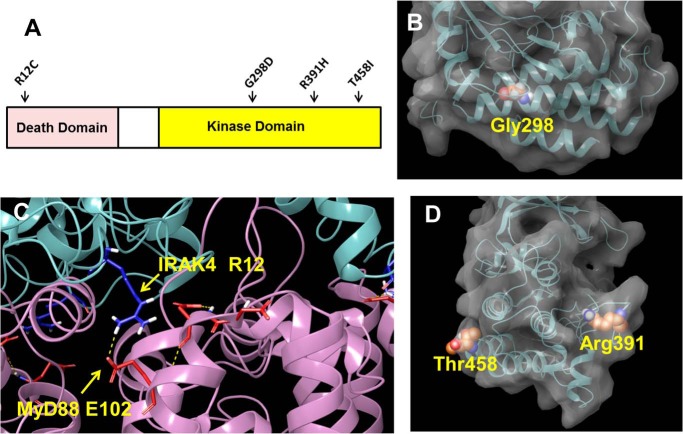
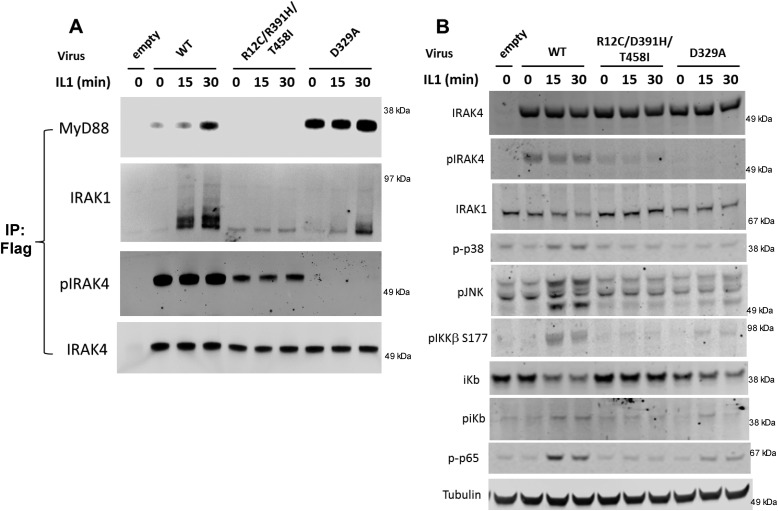
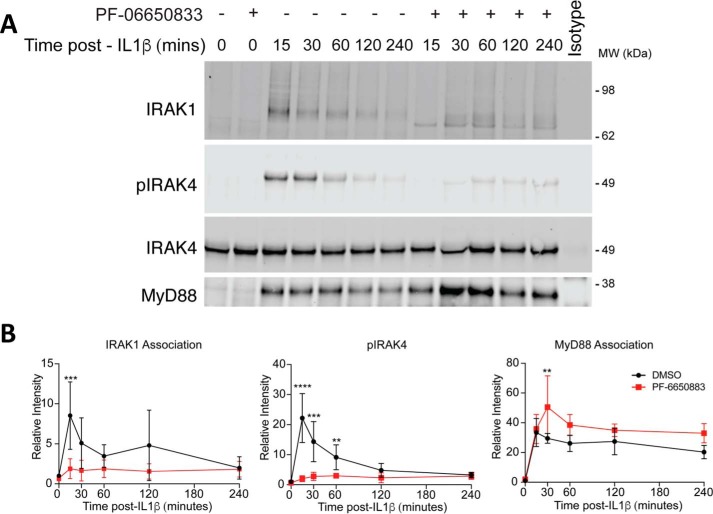
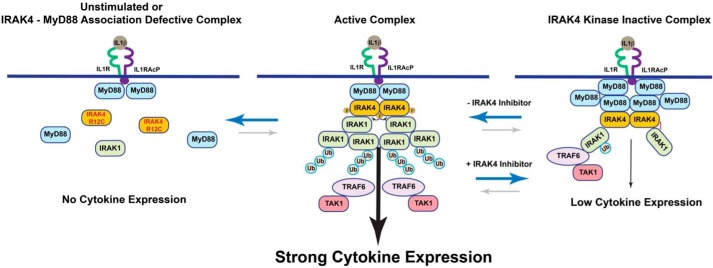
Interleukin-1 receptor (IL1R)-associated kinase 4 (IRAK4) is a central regulator of innate immune signaling, controlling IL1R and Toll-like receptor (TLR)-mediated responses and containing both scaffolding and kinase activities. Humans deficient in IRAK4 activity have autosomal recessive primary immune deficiency (PID). Here, we characterized the molecular mechanism of dysfunction of two IRAK4 PID variants, G298D and the compound variant R12C (R12C/R391H/T458I). Using these variants and the kinase-inactive D329A variant to delineate the contributions of IRAK4's scaffolding and kinase activities to IL1R signaling, we found that the G298D variant is kinase-inactive and expressed at extremely low levels, acting functionally as a null mutation. The R12C compound variant possessed WT kinase activity, but could not interact with myeloid differentiation primary response 88 (MyD88) and IRAK1, causing impairment of IL-1-induced signaling and cytokine production. Quantitation of IL-1 signaling in IRAK4-deficient cells complemented with either WT or the R12C or D329A variant indicated that the loss of MyD88 interaction had a greater impact on IL-1-induced signaling and cytokine expression than the loss of IRAK4 kinase activity. Importantly, kinase-inactive IRAK4 exhibited a greater association with MyD88 and a weaker association with IRAK1 in IRAK4-deficient cells expressing kinase-inactive IRAK4 and in primary cells treated with a selective IRAK4 inhibitor. Loss of IRAK4 kinase activity only partially inhibited IL-1-induced cytokine and NF-κB signaling. Therefore, the IRAK4-MyD88 scaffolding function is essential for IL-1 signaling, but IRAK4 kinase activity can control IL-1 signal strength by modulating the association of IRAK4, MyD88, and IRAK1.
Keywords: Toll/interleukin-1 receptor (TIR); cell signaling; cytokine; inflammation; innate immunity; interleukin 1 (IL-1); interleukin-1 receptor-associated kinase 4 (IRAK4); myeloid differentiation primary response gene 88 (MyD88); protein kinase.
© 2018 De et al.
Conflict of interest statement
S. D., F. K,. and V. R. R. are paid employees of Pfizer Inc. E. K., L. C., and L.-L. L. were paid employees of Pfizer Inc. at the time of this work
Figures



References
-
- Alsina L., Israelsson E., Altman M. C., Dang K. K., Ghandil P., Israel L., von Bernuth H., Baldwin N., Qin H., Jin Z., Banchereau R., Anguiano E., Ionan A., Abel L., Puel A., et al. (2014) A narrow repertoire of transcriptional modules responsive to pyogenic bacteria is impaired in patients carrying loss-of-function mutations in MYD88 or IRAK4. Nat. Immunol. 15, 1134–1142 10.1038/ni.3028 - DOI - PMC - PubMed
-
- Israel L., Wang Y., Bulek K., Della Mina E., Zhang Z., Pedergnana V., Chrabieh M., Lemmens N. A., Sancho-Shimizu V., Descatoire M., Lasseau T., Israelsson E., Lorenzo L., Yun L., Belkadi A., et al. (2017) Human adaptive immunity rescues an inborn error of innate immunity. Cell 168, 789–800.e10 10.1016/j.cell.2017.01.039 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
